病理生理學/休克的病理生理變化
| 醫學電子書 >> 《病理生理學》 >> 休克 >> 休克的病理生理變化 |
| 病理生理學 |
|
|
目錄 |
一、微循環變化
各種休克雖然由於致休克的動因不同,在各自發生髮展過程中各有特點,但微循環障礙(缺血、淤血、播散性血管內凝血)致微循環動脈血灌流不足,重要的生命器官因缺氧而發生功能和代謝障礙,是它們的共同規律。休克時微循環的變化,大致可分為三期,即微循環缺血期、微循環淤血期和微循環凝血期。下面以低血容量性休克為例闡述微循環障礙的發展過程及其發生機理。
低血容量性休克常見於大出血、嚴重的創傷、燒傷和脫水。其微循環變化發展過程比較典型(圖10-1)。
(一)微循環缺血期(缺血性缺氧期)
此期微循環變化的特點是:①微動脈、後微動脈和毛細血管前括約肌收縮,微循環灌流量急劇減少,壓力降低;②微靜脈和小靜脈對兒茶酚胺敏感性較低,收縮較輕;③動靜脈吻合支可能有不同程度的開放,血液從微動脈經動靜脈吻合支直接流入小靜脈。
引起微循環缺血的關鍵性變化是交感神經——腎上腺髓質系統強烈興奮。不同類型的休克可以通過不同機制引起交感——腎上腺髓質性休克和心源性休克時,心輸出量減少和動脈血壓降低可通過竇弓反射使交感——腎上腺髓質系統興奮;在大多數內毒素性休克時,內毒素可直接剌激交感——腎上腺髓質系統使之發生強烈興奮。
交感神經興奮、兒茶酚胺釋放增加對心血管系統的總的效應是使外周總阻力增高和心輸出量增加。但是不同器官血管的反應卻有很大的差別。皮膚、腹腔內臟和腎的血管,由於具有豐富的交感縮血管纖維支配,。而且α受體又佔有優勢,因而在交感神經興奮、兒茶酚胺增多時,這些部位的小動脈、小靜脈、微動脈和毛細血管前括約肌都發生收縮,其中由於微動脈的交感縮血管纖維分布最密,毛細血管前括約肌對兒茶酚胺的反應性最強,因此它們收縮最為強烈。結果是毛細血管前阻力明顯升高,微循環灌流量急劇減少,毛細血管的平均血壓明顯降低,只有少量血液經直捷通路和少數真毛細血管流入微靜脈、小靜脈,組織因而發生嚴重的缺血性缺氧。腦血管的交感縮血管纖維分布最少,α受體密度也低,口徑可無明顯變化。冠狀動脈雖然也有交感神經支配,也有α和β受體,但交感神經興奮和兒茶酚胺增多卻可通過心臟活動加強,代謝水平提高以致擴血管代謝產物特別是腺苷的增多而使冠狀動脈擴張。
交感興奮和血容量的減少還可激活腎素-血管緊張素-醛固酮系統,而血管緊張素Ⅱ有較強的縮血管作用,包括對冠狀動脈的收縮作用。
此外,增多的兒茶酚胺還能剌激血小板產生更多的血栓素A2(thromboxane A2,TXA2),而。TXA2也有強烈的縮血管作用。
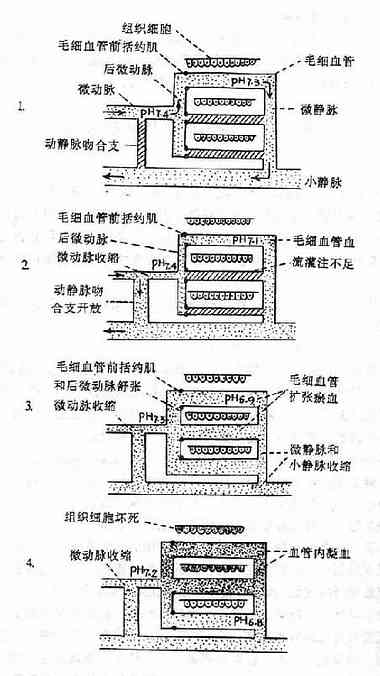
圖10-1 微循環障礙的發展過程模式圖
1.正常情況
⑴動靜脈吻合支是關閉的。
⑵只有20%毛細血管輪流開放,有血液灌流。
⑶毛細血管開放與關閉受毛細血管前括約肌的舒張與收縮的調節。
2.微循環缺血期
⑴交感神經興奮和腎上腺素、去甲腎上腺素分泌增多,小動脈、微動脈、後微動脈,毛細血管前括約肌收縮。
⑵動靜脈吻合支開放,血液由微動脈直接流入小靜脈。
⑶毛細血管血液灌流不足,組織缺氧。
3.微循環淤血期
⑴小動脈和微動脈收縮,動靜脈吻合支仍處於開放狀態,進入毛細血管的血液仍很少。
⑵由於組織缺氧,組織胺、緩激肽、氫離子等舒血管物質增多,後微動脈和毛細血管前括約肌舒張,毛細血管開放,血管容積擴大,進入毛細血管內的血液流動很慢。
⑶由於交感神經興奮,腎上腺素和去甲腎上腺素分泌增多(可能還有組織胺的作用),使微靜脈和小靜脈收縮,毛細血管後阻力增加,結果毛細血管擴張淤血。
4.微循環凝血期
⑴由於組織嚴重缺氧、酸中毒,毛細血管壁受損害和通透性升高,毛細血管內血液濃縮,血流淤滯;另外血凝固性升高,結果在微循環內產生播散性血管內凝血。
⑵由於微血栓形成,更加重組織缺氧和代謝障礙,細胞內溶酶體破裂,組織細胞壞死,引起各器官嚴重功能障礙。
⑶由於凝血,凝血因子(如凝血酶原、纖維蛋白原等)和血小板大量被消耗,纖維蛋白降解產物增多,又使血液凝固性降低;血管壁又受損害,繼而發生廣泛性出血。
而TXA2也有強烈的縮血管作用。
還有,溶酶體水解酶-心肌抑制因子系統在休克Ⅰ期微循環缺血的發生中也起一定的作用。休克時,主要由於胰腺血液灌流量減少所引起的缺血、缺氧和酸中毒可使胰腺外分泌細胞的溶酶體破裂而釋出組織蛋白酶,後者即可分解組織蛋白而生成心肌抑制因子(myocardial depressant factor, MDF)。小分子肽MDF進入血流後,除了引起心肌收縮力減弱、抑制單核吞噬細胞系統的吞噬功能以外,還能使腹腔內髒的小血管收縮,從而進一步加重這些部位微循環的缺血。
本期的主要臨床表現是:皮膚蒼白,四肢厥冷,出冷汗,尿量減少;因為外周阻力增加,收縮壓可以沒有明顯降低,而舒張壓有所升高,脈壓減小,脈搏細速;神志清楚,煩躁不安等。
此期微循環變化具有一定的代償意義。皮膚和腹腔器官等小動脈收縮,既可增加外周阻力,以維持血壓,又可減少這些組織器官的血流量,以保證心腦等重要器官的血液供給;毛細血管前阻力增加,毛細血管流體靜壓降低,促使組織液進入血管,以增加血漿容量;另外,動靜脈吻合支開放,靜脈收縮使靜脈容量縮小(正常約有70%血液在靜脈內),可以加快和增加回心血量,也有利於血壓的維持和心腦的血液供給。但是由於大部分組織器官因微循環動脈血灌流不足而發生缺氧,將導致休克進一步發展。如能及早發現,積極搶救,及時補充血量,降低過劇的應激反應,可以很快改善微循環和恢復血壓,阻止休克進一步惡化,而轉危為安。
這時微循環變化的機理可概括如下(圖10-2):
(二)微循環淤血期(淤血性缺氧期)
在休克的循循環缺血期,如未能及早進行搶救,改善微循環,則因組織持續而嚴重的缺氧,而使局部舒血管物質(如組織胺、激肽、乳酸、腺苷等)增多,後微動脈和毛細血管前括約肌舒張,微循環容量擴大,淤血,發展為休克微循環淤血期。此期微循環變化的特點是:①後微動脈和毛細血管前括約肌舒張(因局部酸中毒,對兒茶酚胺反應性降低),毛細血管大量開放,有的呈不規側囊形擴張(微血池形成),而使微循環容積擴大;②微靜脈和小靜脈對局部酸中毒耐受性較大,兒茶酚胺仍能使其收縮(組織胺還能使肝、肺等微靜脈和小靜脈收縮),毛細血管後阻力增加,而使微循環血流緩慢;③微血管壁通透性升高,血漿滲出,血流淤滯;④由於血液濃縮,血細胞壓積增大,紅細胞聚集,白細胞嵌塞,血小板粘附和聚集等血液流變學的改變,可使微循環血流變慢甚至停止。⑤由於微循環淤血,壓力升高,進入微循環的動脈血更少(此時小動脈和微動脈因交感神經作用仍處於收縮狀態)。由於大量血液淤積在微循環內,回心血量減少,使心輸出量進一步降低,加重休克的發展。
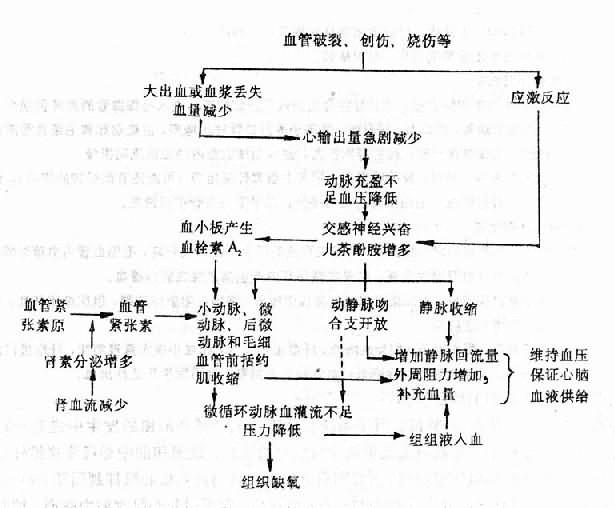
圖10-2 缺血性缺氧期微循環變化機理
由於上述微循環變化,雖然微循環內積有大量血液,但動脈血灌流量將更加減少,病人皮膚顏色由蒼白而逐漸發紺,特別是口辰和指端。因為靜脈迴流量和心輸出量更加減少,病人靜脈萎陷,充盈緩慢;動脈壓明顯降低,脈壓小,脈細速;心腦因血液供給不足,ATP生成減少,而表現為心收縮力減弱(心音低),表情淡漠或神志不清。嚴重的可發生心、腎、肺功能衰竭。這是休克的危急狀態,應立即搶救,補液,解除小血管痙攣,給氧,糾正酸中毒,以疏通微循環和防止播散性血管內凝血。這時微循環變化的機理可概括如下(圖10-3):
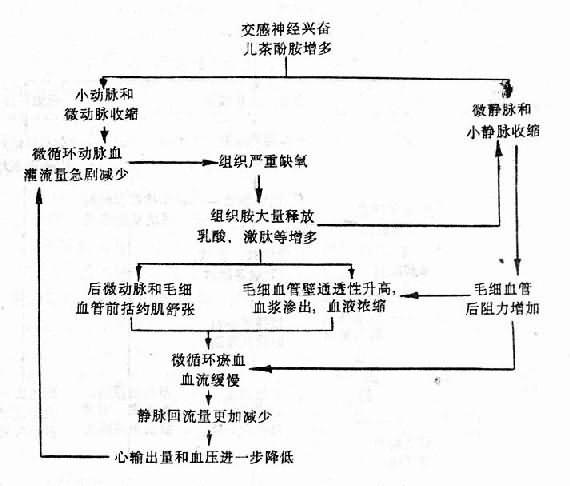
圖10-3 淤血性缺氧期微循環變化機理
(三)微循環凝血期(播散性血管內凝血)
從微循環的淤血期發展為微循環凝血期是休克惡化的表現。其特點是:在微循環淤血的基礎上,於微循環內(特別是毛細血管靜脈端、微靜脈、小靜脈)有纖維蛋白性血栓形成,並常有局灶性或瀰漫性出血;組織細胞因嚴重缺氧而發生變性壞死。
播散性血管內凝血與休克的聯繫極為密切。關於播散性血管內凝血引起的病理變化以及它如何引起休克或加重休克的發展,已在《播散性血管內凝血》一章討論過了,這裡再概要地歸納一下休克如何引起播散性血管內凝血。
1.應激反應使血液凝固性升高。致休克的動因(如創傷、燒傷、出血等)和休克本身都是一種強烈的剌激,可引起應激反應,交感神經興奮和垂體-腎上腺皮質活動加強,使血液內血小板和凝血因子增加,血小板粘附和聚集能力加強,為凝血提供必要的物質基礎。
2.凝血因子的釋放和激活。有的致休克動因(如創傷、燒傷等)本身就能使凝血因子釋放和激活。例如,受損傷的組織可釋放出大量的組織凝血活素,起動外源性凝血過程;大面積燒傷使大量紅細胞破壞,紅細胞膜內的磷脂和紅細胞破壞釋出的ADP,促進凝血過程。
3.微循環障礙,組織缺氧,局部組織胺、激肽、乳酸等增多。這些物質一方面引起毛細血管擴張淤血,通透性升高,血流緩慢,血液濃縮紅細胞粘滯性增加,有利於血栓形成;另一方面損害毛細血管內皮細胞,暴露膠元,激活凝血因子Ⅻ和使血小板粘附與聚集。
4.缺氧使單核吞噬細胞系統功能降低,不能及時清除凝血酶元酶、凝血酶和纖維蛋白。結果在上述因素作用下,而發生播散性血管內凝血(圖10-4)。
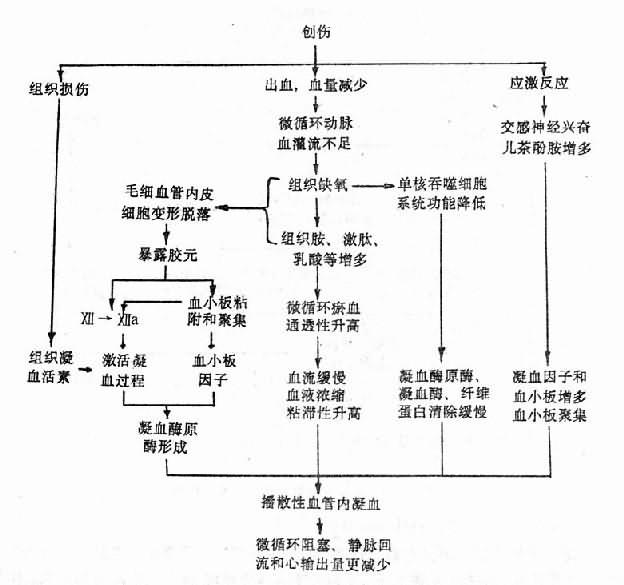
圖10-4 創傷性休克引起播散性血管內凝血的機理
應當指出,在不同類型的休克,播散性血管內凝血形成的早晚可不相同。例如,在燒傷性和創傷性休克時,由於有大量的組織破壞,感染中毒性休克時,由於內毒素對血管內皮的直接損傷,因而都可較早地發生播散性血管內凝血,而在失血性休克等,則播散性血管內凝血發生較晚。
播散性血管內凝血一旦發生,將使微循環障礙更加嚴重,休克病情進一步惡化,這是因為:①廣泛的微血管阻塞進一步加重微循環障礙,使回心血量進一步減少;②凝血物質消耗、繼發纖溶的激活等因素引起出血,從而使血容量減少;③可溶性纖維蛋白多聚體和其裂解產物等都能封閉單核吞噬細胞系統,因而使來自腸道的內毒素不能被充分清除。
由於播散性血管內凝血的發生和微循環淤血的不斷加重,由於血壓降低所致的全身微循環灌流量的嚴重不足,全身性的缺氧和酸中毒也將愈益嚴重;嚴重的酸中毒又可使細胞內的溶酶體膜破裂,釋出的溶酶體酶(如蛋白水解酶等)和某些休克動因(如內毒素等)都可使細胞發生嚴重的乃至不可逆的損害,從而使包括心、腦在內的各重要器官的機能代謝障礙也更加嚴重(詳後),這樣就給治療造成極大的困難,故本期又稱休克難治期。
二、血液流變學的變化
血液流變學(hemorheology)是研究血液流動和變形的科學,或者說是研究血液的流變性、凝固性、血液有形成分(主要是紅細胞)粘彈性以及心血管的粘彈性和變形的科學。物體在一定外力作用下能流動或變形的特性,稱為該物體流變性。一切流體在一定外力作用下,都具有流動性,但流動的難易,則主要取決於流體內部對於流動起阻抗作用的分子之間和顆粒之間的內摩擦力(即流體的粘度)。例如,水的粘度低,容易流動,即流度大;血液的粘度大(紅為蒸餾水的4-5倍),不易流動,即流度小。由於流體的流動是以物體的變形為基礎,所以流體的粘度是映流體流變性的重要指標。
血液是由水、無機鹽、蛋白質、脂類、糖等大小分子所組成的混合液,其中還懸浮著大量具有可塑性的紅細胞,所以血液是一種高濃度的懸濁液。因此能夠影響血液流變性的因素主要有:血細胞壓積(血液粘度隨血細胞的壓積增加而升高)、血細胞的分散程度(血細胞處於分散狀態,血液粘度較低;紅細胞或血小板發生聚集,血液粘度升高)、紅細胞的可塑性(紅細胞可塑性降低,不易變形,血液粘度增加)、血漿內高分子化合物的濃度(血漿粘度大小與其所含蛋白質、脂類、糖等的濃度呈正比)、血管內壁平滑度(血管內皮受損、變形,流經的血液粘度升高)。此外,與血管的長度、口徑、血管壁的彈性和張力也有關係。
休克時血液流變學的主要變化是:
1.血細胞比容血細胞比容的改變與休克的原因和發展階段有關。在低血容量性休克的早期,由於組織間液向血管內轉移,導致血液稀釋,血細胞比容降低,當休克進入微循環淤血期,由於微血管內流體靜壓升高和毛細血管通透性增高,液體乃從毛細血管內外滲至組織間隙,因而血液濃縮,血細胞比容升高。血細胞比容越高,血液粘度越大,血流阻力越大,而血流量則越少,血流更加緩慢。
2.紅細胞變形能力降低,聚集力加強在正常情況下,紅細胞在流經小於其直徑的毛細血管時,可摺疊、彎曲而發生多種變形以減少其寬度,從而得以順利通過。現已證明,休克時紅細胞的變形能力明顯降低,其主要原因是:①休克Ⅱ期時因血液濃縮和組織缺氧所引起的血液滲透壓升高和pH降低,可使紅細胞膜的流動性和可塑性降低並使紅細胞內部的粘度增加;②ATP缺乏(可由缺氧或某些休克動因直接引起)可使紅細胞不能維持正常的功能和結構。結果是由於紅細胞的變形能力降低而難以通過毛細血管,從而導致血流阻力增高。
紅細胞聚集加強,是休克時細胞流變學的重要改變之一。輕者4、5個紅細胞聚集在一起,重者20~30個紅細胞聚集成長鏈或團塊。引起紅細胞聚集的原因是:①血流速度變慢,切變率(shear rate)降低:正常時由於血流速度快和切變率高。可防止紅細胞的聚集,並可促使聚集的紅細胞解聚。休克時隨著血壓下降,血液流速減慢和切變率降低,紅細胞就易於聚集。②紅細胞表面電荷減少:正常紅細胞表面因含有唾液酸的羧基,故都帶有負電荷。紅細胞之間的這種同電荷的排斥力可阻止紅細胞互相靠攏和聚集。休克時,尤其是內毒素性休克時,紅細胞表面負電荷減少,可能是由於血漿中帶正電荷的蛋白質增多,被紅細胞吸附所致,從而使紅細胞彼此靠攏發生聚集。③血細胞比容增加:已如前述,休克時由於血漿外滲,血液濃縮,故血細胞比容增加,這就可以促進紅細胞聚集。④纖維蛋白原濃度增高;纖維蛋白原覆蓋於紅細胞表面,在紅細胞之間可形成有相互聚集作用的「橋力」。休克時由於纖維蛋白原濃度增高,致使「橋力」增大乃至超過負電荷的排斥力。因而就可導致紅細胞的聚集。紅細胞聚集輕則增加血液粘度和血流阻力,重則可引起紅細胞淤滯並阻塞微循環,甚至形成微血栓。
(一)白細胞粘著和嵌塞
正常微循環的血流是紅細胞位於中央的軸流,血漿構成邊流,雖然也可見到少量白細胞附壁滾動,但不發生附壁粘著現象。休克時可見白細胞附著於小靜脈壁,致使血流阻力增高和靜脈迴流障礙。發生白細胞附壁的原因可能與白細胞和管壁之間吸引力增大,休克時血流變慢和切應力(shear stress)下降等因素有關。休克時,還可見到白細胞嵌塞於血管內皮細胞核的隆起處或毛細血管分支處,這可增加血流阻力和加重微循環障礙,而且嵌塞的白細胞還可釋放自由基和溶酶體酶類物質,從而破壞生物膜和引起壞死。休克時白細胞發生嵌塞的原因是:①白細胞的變形能力降低,故不易通過毛細血管而發生嵌塞;②休克時血壓下降,脈壓差減小,動脈血流量減少,驅動白細胞通過毛細血管的力量減弱,因而易於發生白細胞嵌塞。
(二)血小板粘附和聚集
血小板粘附是指血小板和血小板以外的物質相互粘附的現象,血小板聚集則是血小板之間相互發生反應並形成血小板團(或稱血小板聚集物)的過程。粘附一旦開始,聚集過程也隨之發生。在血小板聚集開始時,其表面首先失去光滑性,變得粗糙,形成有突剌的球狀體(或稱聚集型血小板)。在內毒素性、創傷性和燒傷性休克時,血液中這種聚集型血小板的數目增多,而且在微血管中有血小板粘附、聚集和血小板微血栓的形成。這種聚集的血小板不但阻塞微血管,還可釋放多種生物活性物質如兒茶酚胺、TXA2、5-羥色胺等,使局部微血管收縮、通透性增高、血管內皮水腫和血流減少。此外,尚可釋放促凝血的血小板因子(如PF3等),加速凝血過程,形成DIC。
休克時引起血小板粘附和聚集的主要原因是:①血流減慢,血管內皮完整性破壞,內膜下膠原暴露,為血小板粘附提供了基礎;②損傷的內皮組織釋放ADP,發生聚集的血小板可釋放ADP、TXA2以及血小活化因子(PAF),均可觸發並加重血小板的聚集。
(三)血漿粘度增大
休克時,尤其是嚴重創傷或燒傷休克時,一方面由於機體發生應激,使體內合成纖維蛋白原增多;另一方面,在休克的微循環淤血期,毛細血管內的流體靜壓增高,微血管周圍的肥大細胞又因缺氧而釋放組胺並從而使毛細血管通透性增高,液體乃從毛細血管大量外滲至組織間隙,因而血液濃縮,使血漿纖維蛋白原濃度增高,有時纖維蛋白原可高達10g/L(1000mg/dl),故可使血漿粘度增大。這不但影響組織血液流量,並可促進紅細胞的聚集。如當纖維蛋白原的濃度增到5~8g/L(500~800mg/dl)時,由於血漿粘度的增高,紅細胞就發生聚集,形成緡錢狀。
總之,由於發生上述血液流變學的改變,不但會加重微循環障礙和組織的缺血缺氧,還可促進DIC的形成和休克的發展,近年來應用血液稀釋治療休克,其目的在於改善血液流變學,降低血流粘度。這種療法已取得良好的效果。
三、細胞代謝的變化以及功能、結構的損害
休克時細胞的代謝障礙及其功能、結構的損害,既是組織低灌流、微循環流變學改變和/或各種毒性物質作用的結果,又是引起各重要器官功能衰竭和導致不可逆性休克的原因。
(一)休克時細胞的代謝變化
休克時細胞代謝改變比較複雜。由於休克的類型、發展價段以及組織器官的不同,其代謝改變的特點和程度也都有所不同,但共同的重要改變是:
1.糖酵解加強休克時由於組織的低灌流和細胞供氧減少,使有氧氧化受阻,無氧酵解過程加強,從而使乳酸產生增多,而導致酸中毒。但嚴重酸中毒又可抑製糖酵解限速酶如磷酸果糖激酶等的活性,使糖酵解從加強轉入抑制。
2.脂肪代謝障礙正常情況下,脂肪分解代謝中產生的脂肪酸隨血液進入細胞漿後,在脂肪醯輔酶A(脂肪醯CoA)合成酶的作用和ATP的參與下,被活化為水溶性較高的的脂肪醯CoA,後者再經粒線體膜上肉毒鹼脂肪醯轉移酶的作用而進入粒線體中,通過β-氧化生成乙醯輔酶A,最後進入三羧酸循環被徹底氧化。休克時,由於組織細胞的缺血缺氧和酸中毒,使脂肪醯CoA合成酶和肉毒鹼脂肪醯轉移的活性降低,因而脂肪酸的活化和轉移發生障礙;另方面因粒線體獲氧不足和/或某些休克動因(如細菌內毒素)、酸中毒等的直接作用使粒線體呼吸功能被抑制,使轉入粒線體內的脂肪醯CoA不能被氧化分解,結果造成脂肪酸和/或脂肪醯CoA在細胞內蓄積,從而加重細胞的損害。
(二)休克時細胞的損害
休克時細胞的損害首先是生物膜(包括細胞膜、粒線體膜和溶酶體膜等)發生損害。
1.細胞膜的損害 最早的改變是細胞膜通透性增高,從而使細胞內的Na+、水含量增加而K+則向細胞外釋出,細胞膜內外Na+、K+分布的變化,使細胞膜Na+-K+ATP酶活性增高。因而ATP消耗增加,再加上ATP的供應不足和膜上受體腺苷酸環化酶系統受損,結果使控制細胞代謝過程的第二信使-cAMP含量減少,因此細胞的許多代謝過程發生紊亂,例如休克時肌肉細胞對胰島素的反應減弱,使胰島素促進細胞攝取葡萄糖的效應減弱甚至喪失。
休克時引起細胞膜損害的原因是多方面的:
(1)能量代謝障礙休克時因組織細胞的缺血缺氧,一方面ATP生成不足,使細胞膜不能維持正常功能和結構;另一方面脂肪酸氧化受阻,蓄積於細胞內的脂肪酸和脂肪醯CoA與細胞內Na+、K+、Ca+等陽性離子結合形成「皂類」化合物,可直接對膜上脂類起「淨化去垢」的破壞作用。
(2)細胞酸中毒休克時細胞發生酸中毒,除與乳酸等蓄積有關外,還可能與下述因素有關:①細胞低灌流,使產生的CO2不易排出;②ATP分解過程中產生H+(MgATP2-→MgADP-+Pi2-+H+);③胞漿Ca2+增多,可促使Ca2+進入粒線體並與其中的磷酸結合,在結合過程中也產生H+(3Ca2++2HPO42-→Ca3(PO4)2+2H+)。酸中毒可直接或間接破壞膜系統的功能和結構。
(3)氧自由基的產生休克時氧自由基產生增多主要是由於①氧代謝途徑改變:即休克時由於細胞的缺氧和/或內毒素對粒線體呼吸功能的直接抑制,細胞色素氧化酶系統功能失調,以致進入細胞內的氧經單電子還原而形成的氧自由基增多而經4價還原而形成的水減少;②休克時產生大量乳酸、NADH及由ATP分解產生的次黃嘌吟等物質都可提供電子,使氧發生不全性還原而變成氧自由基。另外,休克時因蛋白水解酶活性增高,可催化黃嘌吟脫氫 酶變為黃嘌呤氧化酶,從而使次黃嘌吟變成黃嘌呤和氧自由基。③感染性炎症,活化補體等可激活中性粒細胞和巨噬細胞,使之釋放出氧自由基。
氧自由基可通過膜脂質過氧化反應而破壞生物膜(參閱《缺血與再灌注損傷》)。
此外,溶酶體酶、內毒素等也可破壞細胞膜的功能與結構。
由於細胞膜的完整性在維持細胞的生命活動中起著重要作用。故當膜完整性破壞時,即意味著細胞不可逆性損傷的開始。
2.粒線體損害休克時粒線體最早出現的損害是其呼吸功能和ATP合成受抑制,粒線體ATP酶活性降低。此後發生超微結構的改變,如基質顆粒減少或消失;繼之,基質電子密度增加、嵴內腔擴張,隨後,嵴明顯腫脹,終至破壞。
關於休克時粒線體損害的原因尚不完全清楚。缺氧可減少粒線體合成ATP,但除非在嚴重缺氧和伴有缺血時,並不引起粒線體膜的明顯損害。目前認為,粒線體損害可能與下列因素有關:①內毒素等毒性物質及酸中毒對粒線體各種呼吸酶的直接抑制;②缺血導致粒線體合成ATP的輔助因子(如NAD、CoA和腺苷等)不足和細胞內環境(pH、離子)的改變。③前述的氧自由基對粒線體膜磷脂的過氧化作用等。
粒線體是維持細胞生命活動的「能源供應站」。粒線體損害時,由於氧化磷酸化障礙,產能減少乃至終止,故必然導致細胞損害和死亡。
3.溶酶體破裂溶酶體含有多種水解酶,如組織蛋白酶、多肽酶、磷酸酶等,但在未釋放之前都處於無活性狀態。一旦釋放出來後,它們即轉為活性狀態而可溶解和消化細胞內、外的各種大分子物質,尤其是蛋白類物質。已證明,休克早期,肝、脾、腸等細胞即出現溶酶體腫大,顆粒喪失和酶釋放增加;內毒素休克動物血液和淋巴中水解酶濃度增高,且與休克嚴重程度呈正相關。給動物注射溶酶體或溶酶體酶,可產生類似休克的各種病理生理改變。
休克時導致溶酶體破裂的主要原因是:①組織的缺血、缺氧、酸中毒以及內毒素對溶酶體膜的直接破壞;②氧自由基對溶酶體膜磷脂的過氧化作用;③血漿補體被激活產生C5a,後者可剌激中性粒細胞釋放溶酶體酶。釋放的溶酶體酶又可通過多種途徑參與休克的發生、發展和細胞的損害,例如:a.釋放的組織蛋白酶使蛋白質水解,這不但可以破壞蛋白酶的活性,甚至還可使細胞自溶壞死,而且所產生的多肽類活性物質,還能加重微循環障礙;b.破壞生物膜的完性;c.直接損害血管內皮和血管平滑肌細胞,從而導致血液外滲、出血和血小板的粘附、聚集以及DIC形成;d.激活補體系統產生C5a,後者再進一步促使溶酶體酶的釋放。現已證明,休克時使用溶酶體膜穩定藥可防止或減輕溶酶體膜的破裂。
總之,休克時生物的損害被認為是細胞發生損害的開始,而細胞的損害又是各臟器功能衰竭的共同機制。
四、器官功能的改變
休克時各器官功能都可發生改變,其中主要是中樞神經系統、心、腎、肺、胃腸及肝臟等重要器官的功能障礙。
(一)中樞神經系統功能的改變
休克早期,如果能通過代償性調節維持腦的血液供給,除因應激反應而有興奮性升高外,一般沒有明顯的腦的功能障礙。休克進一步發展,心輸出量減少和血壓降低,不能維持腦的血液供給,則發生缺氧。嚴重的缺氧和酸中毒還能使腦的微循環血管內皮細胞和小血管周圍的神經膠質細胞腫脹,致腦微循環狹窄或阻塞,動脈血灌流更加減少。在微循環凝血期,腦循環內可以有血栓形成和出血。大腦皮層對缺氧極為敏感,當缺氧逐漸加重,將由興奮轉為抑制(表情淡漠),甚至發生驚厥和昏迷。皮層下中樞因嚴重缺氧也可發生抑制,呼吸中樞和心血管運動中樞興奮性降低(詳見《缺氧》)。
(二)心臟功能的改變
除心源性休克伴有原發性心功能障礙外,其它各類型休克也都可引起心功能的改變。一般而言,休克的早期可出現心的代償性加強,此後心臟的活動即逐漸被抑制,甚至可出現心力衰竭,其主要機制是:
1.冠脈血流量減少和心肌耗氧量增加 由於休克時血壓降低以及心率加快所引起的心室舒張期縮短,可使冠脈灌流量減少和心肌供血不足;同進因交感-兒茶酚胺系統興奮使心率加快、心縮加強,導致心肌耗氧量增加,因而更加重了心臟缺氧。結果心肌因能量不足和酸中毒而使舒縮功能發生障礙,並從而引起心力衰竭,對於原來就有冠狀動脈供血不良者,尤其容易出現心力衰竭。
2.酸中毒和高鉀血症 酸中毒可通過多種機制影響心臟舒縮功能:①抑制肌膜的Ca2+內流;②H+和Ca2+競爭結合肌鈣蛋白;③抑制肌漿網對Ca2+的攝取和釋放;④抑制肌球蛋白ATP酶的活性。此外,酸中毒還可通過抑制心肌細胞能量代謝酶的活性、促使生物膜的破壞以及誘發心律失常等多種途徑來抑制心肌的舒縮功能,並從而促使心力衷竭的發生。
休克時,組織細胞的破壞可釋出大量K+,腎功能的障礙又使K+的排出減少,因而總是伴有高鉀血症。高血鉀可抑制動作電位復極化2期中Ca2+的內流,從而使心肌興奮-收縮偶聯減弱。
此外,心肌內DIC形成,內毒素對心肌的直接作用等等。都可以促使心肌力衰竭的發生。一旦發生了心力衰竭,將迅速促使休克進一步惡化,並給輸液擴容造成一定困難。
3.心肌抑制因子的作用如前文所述,休克時的缺血、缺氧等可使胰腺產生心肌抑制因子(MDF),MDF能使心肌收縮力減弱,從而有助於心力衰竭的發生。
(三)腎功能的改變
腎功能的改變在休克早期就可發生,這時發生的是功能性的急性腎功能衰竭,因為它還不伴有腎小管的壞死。其主要臨床表現為少尿(<400ml/d)或無尿(<100ml/d),其發生的主要機制如下:
1.腎小球濾過率減少在休克早期,有效循環血量的減少不僅能直接使腎血流量不足,而且還可通過腎素-血管緊張素系統和交感-兒茶酚胺系統的激活而使腎血管收縮,因而使腎血流量更加減少,結果是腎小球濾過壓降低,腎小球濾過率減少。
2.腎小管對鈉、水重吸收加強在休克早期,腎小管上皮細胞雖然已經發生缺血,但是因為持續時間不久,故這些細胞仍能保持其正常的重吸收功能,加之此時醛固酮和抗利尿激素分泌增多,所以腎小管對鈉水的重吸收加強。腎小球濾過率的減少和腎小管重吸收的增強就可導致少尿或無尿。但此時腎功能的變化是可逆的。一旦休克逆轉,血壓恢復,腎血流量和腎功能即可恢復正常,尿量也將隨之而恢復正常。故尿量變化是臨床判斷休克預後和療效的重要指標。
當休克持續時間較長時,可引起急性腎小管壞死,發生器質性的腎功能衰竭。此時即使腎血流量隨著休克的好轉而恢復,患者的尿量也難以在短期內恢復正常。腎功能的這些改變,將導致嚴重的內環境紊亂,包括高鉀血症、氮質血症和酸中毒等。這樣就會使休克進一步惡化,故許多休克患者,尤其是老年患者常死於急性腎功能衰竭。
(四)肺功能的改變
隨著休克的發展,肺功能也發生不同程度的改變:在休克早期,由於呼吸中樞興奮,故呼吸加快加深,通氣過度,從而甚至可以導致低碳酸血症和呼吸性鹼中毒;繼之,由於交感-兒茶酚胺系統興奮和其他血管活性物質的作用,可使肺血管阻力升高;如果肺低灌流狀態持續較久,則可引起肺淤血、水腫、出血、局限性肺不張、微循環血栓形成和栓塞以及肺泡內透明膜形成等重要病理改變,此即所謂休克肺(shock lung)的病理學基礎(參閱第十三章)。
上述休克肺的病理變化,有的影響肺的通氣功能。有的妨礙氣體彌散,有的改變肺泡通氣量/血流量的比例,造成死腔樣通氣和/或功能性分流,從而可以導致呼吸衰竭甚至死亡。休克肺是休剋死亡的重要原因之一,約有1/3的休克患者死於休克肺。
(五)肝和胃腸功能的改變
1.肝功能的改變休克時常有肝功障礙,其主要原因有:①低血壓和有效循環血量減少可使肝動脈血液灌流量減少,從而引起肝細胞缺血缺氧,嚴重者可導致肝小葉中央部分肝細胞壞死;②休克時由於腹腔內髒的血管收縮,致使門脈血流量急劇減少。肝約有一半以上血液來自門脈,故門脈血流量減少,也將加重肝細胞的缺血性損害;③肝內微循環障礙和DIC形成,更可引起肝細胞缺血缺氧;④在腸道產生的毒性物質經門脈進入肝,加之肝本身毒性代謝產物的蓄積對肝細胞都有直接損害作用。
肝功能障礙又可通過下列機制加重休克:①肝代謝障礙:肝對糖和乳酸的利用障礙,一方面可促使乳酸蓄積並從而引起酸中毒;另方面又不能為各重要臟器提供充足的葡萄糖。蛋白質和凝血因子合成障礙,可引起低蛋白血症和出血。②肝的生物轉化作用(解毒功能)減弱:可增加休克時感染與中毒的危險。
2.胃腸功能的改變 休克早期就有胃腸功能的改變。開始時是因微小管痙攣而發生缺血,繼而可轉變為淤血,腸壁因而發生水腫甚至壞死。此外,胃腸的缺血缺氧,還可使消化液分泌抑制,胃腸運動減弱。有時可由於胃腸肽和粘蛋白對胃腸粘膜的保護作用減弱,而使胃腸粘膜糜爛或形成應激性潰瘍。
由於胃腸上述改變,可通過下列機制促使休克惡化:①腸道粘膜屏障功能減弱或破壞,致使腸道細菌毒素被吸收入血,加之肝的生物轉化作用減弱,故易引起機體中毒和感染。②胃微循環淤血,血管內液體外滲,加之胃腸粘膜糜爛壞死和DIC的形成都可導致胃腸道出血,從而使血容量進一步減少。③胃腸道缺血、缺氧,可剌激肥大細胞釋放組胺等血管活性物質,因而微循環障礙進一步加劇。
(六)多器官功能衰竭
多器官功能衰竭(mrltiple organ failure MOF)是指心、腦、肺、腎、肝、胃腸、胰腺及血液等器官中,在24小時內有兩個或兩個以上的器官相繼或同時發生功能衰竭。MOF又稱多系統功能衰竭或綜合器官衰竭。休克的晚期常出現MOF。MOF是致死的重要原因,而且衰竭的器官越多,病死率也就越高。如有三個器官發生功能衰竭時,病死率可高達80%以上。
MOF在臨床上有兩種表現形式,一是創傷和休克直接引起的速髮型,又稱單相型,發生迅速,發病後很快出現肝、腎和呼吸功能障礙,在短期內或則死亡,或則恢復;二是創傷、休克後繼發感染所致的遲髮型,又稱雙相型。此型患者往往有一個相對穩定的間歇期,多在敗血症發生後才相繼出現多器官功能衰竭。引起MOF的主要原因是:①重症感染:約有70~80%的MOF是在重症感染的基礎上發生的;②休克時組織較長時間的低灌流和交感神經的高反應;③非感染性的嚴重病變如急性胰腺炎、廣泛性組織損傷等。尤其是當機體的免疫功能和單核吞噬細胞系統功能減弱時,或者是治療不當或延誤時,如未及時糾正組織低灌流和酸鹼平衡紊亂、過多過快輸液、大量輸血或過量應用鎮靜劑、麻醉劑等情況下,更易引起MOF。
MOF的發病機制尚不很清楚,現認為MOF的發生是多因素參與作用的結果,其中休克時組織低灌流所致的組織缺血缺氧、代謝障礙和酸中毒都起著重要作用;在感染或感染中毒性休克時,細菌內毒素在MOF的發生機制中被認為是起著關鍵的作用。這是因為內毒素不但能直接或間接(如激活補體和凝血系統)損害各器官的功能,而且還可通過激活補體而使中性粒細胞聚集和激活,從而使中粒細胞①釋放各種水解酶,包括酸性和鹼性蛋白酶,這些酶不但能破壞結構蛋白(如彈性蛋白酶破壞彈性纖維,膠原酶破壞膠原纖維等),而且還可分解血漿蛋白,激活凝血系統並從而導致彌散性血管內凝血;②產生並釋放活性氧和脂類代謝產物(如白三烯等),這些物質又可破壞生物膜和/或增高血管通透性,加重微循環障礙。在一般情況下,中性粒細胞向感染和損傷處趨化和聚集,是炎性的正常反應,但在休克或嚴重感染時,由於機體免疫功能降低等原因,對這種炎性反應失去控制,從而使中性粒細胞釋放的上述各種毒性物質得以廣泛地破壞各器官細胞的結構和功能。故有人認為MOF是一種非特異性失控的全身性炎性反應。此外,兒茶酚胺-腺苷酸環化酶-cAMP系統異常也可能起著重要的作用,在休克時一方面因為細胞的缺血缺氧,使膜功能異常,腺苷酸環化酶系統的受體受損,對兒茶酚胺的反應減弱;另方面由於組織ATP含量降低,缺乏產生cAMP的底物,結果使細胞內cAMP水平下降,進而影響細胞內的許多代謝過程和功能。
| 關於「病理生理學/休克的病理生理變化」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |