病理學/自身免疫性疾病的類型和舉例
| 醫學電子書 >> 《病理學》 >> 免疫病理 >> 自身免疫性疾病 >> 自身免疫性疾病的類型和舉例 |
| 病理學 |
|
|
|
自身免疫性疾病可分為二大類:
(一)器官特異性自身免疫病
組織器官的病理損害和功 能障礙僅限於抗體或致敏淋巴細胞所針對的某一器官。主要有慢性淋巴性甲狀腺炎、甲狀腺功能亢進、胰島素依賴型糖尿病、重症肌無力、慢性潰瘍性結腸炎、惡性 貧血伴慢性萎縮性胃炎、肺出血腎炎症候群(goodpasture syndrome)、尋常天皰瘡、類天皰瘡、原發性膽汁性肝硬變、多發性腦脊髓硬化症、急性特發性多神經炎等,其中常見者將分別於各系統疾病中敘述。
(二)系統性自身免疫病
由 於抗原抗體複合物廣泛沉積於血管壁等原因導致全身多器官損害,稱系統性自身疫病。習慣上又稱之為膠原病或結締組織病,這是由於免疫損傷導致血管壁及間質的 纖維素樣壞死性炎及隨後產生多器官的膠原纖維增生所致。事實上無論從超微結構及生化代謝看,膠原纖維大多並無原發性改變,以下簡述幾種常見的系統性自身免 疫病。
1.系統性紅斑狼瘡(systemic lupus erythematosus, SLE) 是一種比較常見的系統性自身免疫病,具有以抗核抗體為主的多種自身抗體和廣泛的小動脈病變及多系統的受累。臨床表現主要有發熱,皮損(如面部蝶 形紅斑)及關節、腎、肝、心漿膜等損害,以及全血細胞的減少。多見於年輕婦女,男女比為1:6~9,病程遷延反覆,預後差。
病因與發病機制 本病的病因和發病機制不明,目前的研究主要集中在以下三個面。
(1)免疫因素:患者體內有多種自身抗體形成,提示B細胞活動亢進是本病的發病基礎。周圍血中B細胞體外培養實驗結果發現其增殖能力較正常強8~10倍。
(2) 遺傳因素:遺傳因素與本病的關係表現為:①在純合子雙胎中有很高(69%)的一致性,②SLE患者家屬成員中發病的可能性明顯增加,③北美白人中SLe 與HLA DR2、DR3有關。這可能是由於位於HLA D區的免疫反應基因(Ir)對抗原(包括自身抗原)所激發的免疫反應的程度有調節作用的緣故。
(3) 其他:非遺傳因素在啟動自身免疫反應中亦起著一定的作用。這些因素包括:①藥物:鹽酸肼苯噠嗪(hydralazine)、普魯卡因醯胺 (procainamide)等可引起SLE樣反應。但停藥後常可自愈;②病毒:在實驗動物NZB和NZB/WF1小鼠中的自發性SLE樣病中發現C型病 毒感染,在腎小球中可檢出病毒抗原-抗體複合物。但在SLE病中病毒因素尚未能充分得到證實;③性激素對SLE的發生有重要影響,其中雄激素似有保護作 用,而雌激素則似有助長作用,故患者以女性為多,特別多發生在生育年齡,病情在月經和妊娠期加重。
自身抗體及組織損害機制本病患者體內有多 種自身抗體,95%以上病人抗核抗體陽性,可出現抗DNA(雙股、單股)、抗組蛋白、抗RNA-非組蛋白、抗核糖核蛋白(主要為Smith抗原)、抗粒細 胞、抗血小板、抗平滑肌等抗體,其中抗雙股DNA和抗Smith抗原具相對特異性,陽性率分別為60%和30%,而在其他結締組織病的陽性率,均低於 5%。
抗核抗體並無細胞毒性,但能攻擊變性或胞膜受損的粒細胞,一旦它與細胞核接觸,即可使胞核腫脹,呈均質狀一片,並被擠出胞體,形成狼 瘡(LE)小體,LE小體對中性粒細胞、巨噬細胞有趨化性(圖4-8),在補體存在時可促進細胞的吞噬作用。吞噬了LE小體的細胞為狼瘡細胞(圖 4-9)。在組織中,LE小體呈圓或橢圓形,HE染色時蘇木素著色而藍染,故又稱蘇木素小體,主要見於腎小球或腎間質。一般僅在20%的患者可檢見蘇木素 小體,為診斷SLE的特徵性依據。
SLE的組織損害與自身抗體的存在有關,多數內臟病變是免疫複合物所介導(Ⅲ型變態反應),其中主要為DNA-抗DNA複合物所致的血管和腎小球病變,其次為特異性抗紅細胞、粒細胞、血小板自身抗體經Ⅱ型變態反應導致相應血細胞的損害和溶解,引起全貧血。
病 變急性壞死性小動脈、細動脈炎是本病的基礎病變,幾乎存在於所有患者並累及全身各器官。活動期病變以纖維素樣壞死為主。慢性期血管壁纖維化明顯,管腔狹 窄,血管周圍有淋巴細胞浸潤伴水腫及基質增加。有時血管外膜纖維母細胞增生明顯,膠原纖維增多,形成洋蔥皮樣結構,以脾中央動脈的變化最為突出。應用免疫 組織化學方法可證實受累的血管壁中有免疫球蛋白、補體、纖維蛋白、DNA等存在,提示有抗原-抗體複合物機制的參與。
(1)腎:腎功能衰竭 是SLE的主要死亡原因。SLE病人幾乎均有不同程度的腎損害,約60%病便以狼瘡性腎炎為主要表現.常見的類型有系膜增生型(10%~15%)、局灶增 生型(10%~15%)、瀰漫增生型(40%~50%)和膜型(10%~20%)。各型狼瘡性腎炎的病變,類同於相應的原發性腎小球腎炎,各型病變間常有 交叉,因此腎小球的病變呈多樣性,晚期可出現典型的硬化性腎炎的表現。腎炎病變的發生主要基於腎小球中免疫複合物的沉積,可位於系膜區、內皮下和上皮下。 其中瀰漫增生型狼瘡性腎炎中內皮下大量免疫複合物的沉積,是SLE急性期的特徵性病變。在瀰漫增生型及膜型病例中,約半數病例在間質及腎小管基膜上亦有免 疫複合物沉積,因此腎小球病變和間質的炎症反應在狼瘡性腎炎中十分明顯(圖4-10)。蘇木素小體的出現有明確的診斷意義。

圖4-8 紅斑性狼瘡花形細胞簇
游離的LE小體被多個粒細胞包圍,形成花形細胞族(采自 Dubois)
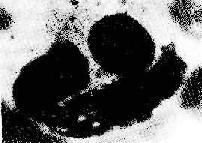
圖4-9 狼瘡細胞
胞漿內吞噬了兩個LE小體,胞核被擠在一邊
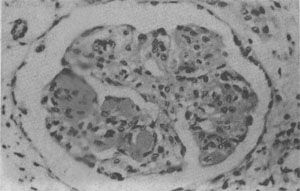
圖4-10 狼瘡性腎小球腎炎
腎小球毛細血管叢節段性纖維素樣壞死,伴系膜細胞增生;間質炎細胞浸潤
(2) 皮膚:約40%的SLE病人有明顯皮膚損害,以面部蝶形紅斑最為典型,亦可累及軀幹和四肢。鏡下,表皮常有萎縮、角化過度、毛囊角質栓形成、基底細胞液 化,表皮和真皮交界處水腫,基度膜、小動脈壁和真皮的膠原纖維可發生纖維素樣壞死,血管周圍常有淋巴細胞浸潤。免疫熒光證實真皮與表皮交界處有IgG、 IgM及C3的沉積,形成顆粒或團塊狀的熒光帶即「狼瘡帶」,可能是壞死上皮細胞釋出之抗原與血循環中彌散出來的抗核抗體等自身抗體形成的免疫複合物。狼 瘡帶的出現對本病有診斷意義。
(3)心:大約半數病例有心臟受累,心瓣膜非細菌性疣贅性心內膜炎(nonbacterial verrucous endocarditis或Libman-Sach endocarditis)最為典型,敖生物常累及二尖瓣或三尖瓣,其特點為:大小自1mm至 3~4mm,數目單個或多個不等,分布極不規則,可累及瓣膜之前後面或心腔之內膜或腱索(圖4-11)。鏡下,贅生物由纖維蛋白和壞死碎屑及炎症細胞構 成,根部基質發生纖維素樣壞死,伴炎細胞浸潤,後期發生機化。
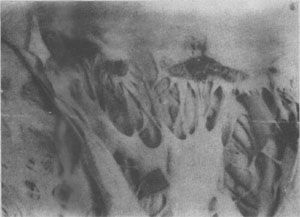
圖4-11 紅斑性狼瘡的心瓣膜疣狀心內膜炎
二尖瓣上有疣狀贅生物形成
(4)關節:90%以上的病例有不同程度的關節受累。滑膜充血水腫,有較多單個核細胞浸潤。於緊接上皮淺表部位的結締組織可出現灶性纖維素樣壞死,但很少侵犯關節軟骨等深部組織,因此極少引起關節畸形。
(5)肝:約25%的病例可出現肝損害,稱狼瘡性肝炎。可表現為匯管區及匯管區周圍的單個核細胞浸潤及附近肝細胞的碎屑狀壞死等慢性活動性肝炎的典型病變。亦可僅有少量散在分布的小灶性壞死等輕微病變。
(6)脾:體積略增大,包膜增厚,濾泡增生頗常見。紅髓中有多量漿細胞,內含IgG、IgM,最突出的變化是小動脈周圍纖維化,形成洋蔥皮樣結構(圖4-12)。
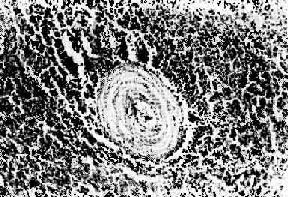
圖4-12 紅斑性狼瘡之脾病變
脾小體中央細動脈壁呈洋蔥皮樣結構
(7)淋巴結:全身淋巴結均有不同程度的腫大,竇內皮增生。其中較多的漿細胞,小血管變化與脾所見相同。
2.口眼乾燥症候群 口眼乾燥症候群(Sjǒgren syndrome)臨床上表現為眼乾、口乾等特徵,乃唾液腺、淚腺受免疫損傷所致。本病可單獨存在,也可與其他自身免疫病同時存在,後者最常見的是類風濕 性關節炎、SLE等。病變主要累及唾液腺及淚腺,其他外分泌腺包括呼吸道、消化道腺體也可受累。
唾液腺的組織學病變主要表現為腺管周圍大量 炎細胞浸潤,主要是淋巴細胞和漿細胞,有時可形成淋巴濾泡並有生髮中心形成。伴腺管上皮增生,引起管腔阻塞。病變晚期腺泡萎縮、纖維化,為脂肪組織所替 代。個別病例浸潤的淋巴細胞形成淋巴瘤樣結構。由於唾液腺的破壞而引起口腔粘膜乾裂及潰瘍形成。
淚腺的類似病變可導致角膜上皮乾燥、炎症及潰瘍形成。呼吸道、消化道受累可導致相應的鼻炎、喉炎、支氣管炎、肺炎及萎縮性胃炎。在腎可發生間質性腎炎,腎小管周圍大量單個核細胞浸潤,導致腎小管萎縮、纖維化,因腎小管功能損害而引起腎小管性酸中毒、磷酸鹽尿等頗常見。
淋巴結腫大並有增生性變化,核分裂多,故又名假性淋巴瘤。值得提出的是本病患者發生惡生淋巴瘤的機會較正常人高40倍。
發 病機制 本病的發病機制尚不清楚。由於常伴發SLE和類風濕性關節炎,提示本病的發生與免疫性損傷有關。患者B細胞功能過度,表現為多克隆高球蛋白血症和類風濕因 子(RF)、抗核抗體、冷球蛋白及抗唾液腺抗體的形成。近年來並發現兩種特徵性抗核糖核蛋白成分的自身抗體,分別命名為抗SS-B和SS-A,在本病有很 高的陽性率(60%、70%),對本病的診斷有參考價值。病灶處有大量B及T細胞浸潤,後者大部分為T輔助細胞,也有一部分為T殺傷細胞,提示亦有細胞免 疫機制的參與。
3.類風濕性關節炎 詳見骨關節疾病章。
4.硬皮病 硬皮病(scleroderma)又名進行性系統性硬化症(progressive systemic sclerosis),以全身許多器官間質過度纖維化為其特徵。95%以上的患者均有皮膚受累的表現;但橫紋肌及許多器官(消化道、肺、腎、心等)受累是 本病主要損害所在,病變嚴重者可導致器官功能衰竭,威脅生命。
病因和發病機制本病病因不明,其發病可能與以下因素有關:
(1)膠原合成增加:體外培養證實,患者纖維母細胞合成膠原的能力明顯高於正常人,合成超過降解,導致大量膠原纖維的積集;
(2)Ⅳ型變態反應:在皮膚病變中有T細胞浸潤,所分泌的淋巴因子及其刺激巨噬細胞分泌的因子可刺激纖維母細胞大量合成膠原;
(3)自身抗體:50%患者有輕度高丙種球蛋白血症及多種自身抗體,包括RF,抗平滑肌抗體,抗核抗體等,可能由於抗原抗體免疫複合物的沉積或內皮細胞毒的作用,造成小血管內皮細胞損傷、血栓形成、管壁纖維化、管腔狹窄,導致組織缺氧而引起纖維間質增生。
【病變】
(1) 皮膚:病變由指端開始,向心性發展,累及前臂、肩、頸、臉,使關節活動受限。早期受累的皮膚發生水腫,質韌。鏡下,主要表現為小血管周圍淋巴細胞浸潤,毛 細血管內皮細胞腫脹、基膜增厚、管腔部分阻塞,間質水腫,膠原纖維腫脹,嗜酸性增強。隨著病變的發展,真皮中膠原纖維明顯增加,並與皮下組織緊密結合,表 皮萎縮變平,黑色素增加,釘突和附屬器萎縮消失,小血管增厚、玻璃樣變。晚期手指細而呈爪狀、關節活動受限,有時指端壞死甚或脫落,面部無表情呈假面具 狀。
(2)消化道:約有1/2患者消化道受累,粘膜上皮萎縮,固有層、粘膜下層、肌層為大量膠原纖維所取代,血管周單個核細胞浸潤。病變以食管下2/3段最嚴重,管腔狹窄,缺乏彈性。小腸、結腸也可受累。臨床上出現吞咽困難、消化不良等症状。
(3)腎:葉間小動脈病變最為突出,表現為內膜粘液樣變性,伴內皮細胞增生及隨後的管壁纖維化、管腔明顯狹窄,部分病例並有細動脈纖維素樣壞死。臨床上可出現高血壓,與惡性高血壓腎病變難以區別。約50%患者死於腎功能衰竭。
(4)肺:瀰漫性間質纖維化,肺泡擴張、胞泡隔斷裂,形成囊樣空腔,本病是造成蜂窩肺的重要原因之一。
5.結節性多動脈炎 結節性多動脈炎(polyarteritis nodosa)是全身動脈系統的疾病,表現為中小動脈壁的壞死性炎症。患者以青年人為多,有時也可發生在兒童及老人、男女之比為2~3:1。
病變各系統或器官的中小動脈均可受累,其中以腎(85%)、心(75%)、肝(65%)、消化道(50%)最為常見。此外,胰、睾丸、骨骼肌、神經系統和皮膚也可受累。
病 變多呈節段性,以血管分叉處最為常見。內眼觀,病灶處形成直徑約2~4mm的灰白色小結節,結節之間的血管壁外觀正常。鏡下,急性期表現為急性壞死炎症, 病變從內膜和中膜內層開始,擴展至管壁全層及外膜周圍,纖維素樣壞死頗為顯著,伴炎細胞浸潤(圖4-13)尤以嗜酸性及中性粒細胞為多,繼而有血栓形成。 以後的進展是纖維增生,管壁呈結節性增厚,管腔機化阻塞和明顯的動脈周圍纖維化。值得注意的是早期炎性壞死變化及後期膠原化可同時存在。病變的主要後果是 缺血性損害和梗死形成。
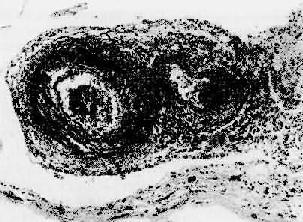
圖4-13 結節性多動脈炎
兩個動脈壁的各層都有炎性細胞浸潤,外膜尤為顯著。中膜發生纖維素樣壞死
本病病變分布廣泛,臨床表現變異多端,患者常有低熱、乏力、粒細胞增多以及多系統受累的症状,如血尿、腎功能衰竭、高血壓、腹痛、腹瀉、黑糞及周圍神經炎等。病程快慢不一,經免疫抑制治療,55%患者可存活。
病因與發病機制 病因和發病機制不明,動物實驗提示,體液因素在本病的發生中起著重要作用。免疫熒光技術證實,人結節性多動脈炎血管壁中有免疫球蛋白和補體,有些還有HBsAg,約50%患者血清HBsAg或抗HBs陽性。
6.Wegener肉芽腫病 Wegener肉芽腫病是一種少見病,具有以下特點:①小血管急性壞死性脈管炎,可累有各器官的血管,以呼吸道、腎、脾最常受累。表現為小動脈、小靜脈管 壁的纖維素樣壞死,伴瀰漫性中性和嗜酸性粒細胞浸潤;②呼吸道肉芽腫性壞死性病變,可累及口、鼻腔、鼻旁竇、喉、氣管、支氣管和肺。病變為由大量積集的單 核巨噬細胞、淋巴細胞以及少量多核巨細胞、類上皮細胞、纖維母細胞組成的肉芽腫,中央可陷於成片凝固性壞死。肉眼常形成明顯的腫塊,表面則因壞死潰破而有 潰瘍形成;③壞死性腎小球腎炎,表現為在局灶性或瀰漫增生性腎小球腎炎的基礎上,有節段性毛細血管袢的纖維素樣壞死,血栓形成,如未經治療可發展為快速進 行性腎炎,病程兇險,出現進行性腎功能衰竭。
本病的病因不明,由於有明顯的血管炎,並於局部可檢得免疫球蛋白和補體,提示其發病與Ⅲ型變態反應有關。但呼吸道出現的肉芽腫和壞死性病變,又提示可能與Ⅳ型變態反應有關,臨床上應用細胞毒藥物大多能使本病緩解。
| 關於「病理學/自身免疫性疾病的類型和舉例」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |