手、足部厚壁水皰
| A+醫學百科 >> 手、足部厚壁水皰 |
大皰性表皮鬆解可表現為局限性大皰性表皮鬆解症,始於兒童時期或更晚,是最常見的一型。也可以到成人時才出現,表現為在高強度運動後出現手、足部厚壁水皰。常見手足多汗。足部的水皰常繼發感染。
目錄 |
手、足部厚壁水皰的原因
(一)發病原因
大皰性表皮鬆解症依其透射電鏡下水皰形成的水平可分為3大類(見表1)。真皮-表皮交界區內編碼蛋白的不同基因突變提供了臨床上不同亞型間表現不同的分子基礎。單純型大皰性表皮鬆解症的表皮鬆解水平在基底細胞層,是基底細胞角質蛋白基因KRT5和KRTl4突變的結果。交界型大皰性表皮鬆解症的組織松解發生在真皮表皮基底膜的透明帶水平,超微結構顯示半橋粒錨細絲複合體異常,其編碼錨細絲蛋白.層粘連蛋白5(1aminin)的3個肽α3、β3和γ2的基因發生特異突變。另外,在交界型大皰性表皮鬆解症的亞型中發現編碼半橋粒組成成分的基因突變,包括編碼α6β4整合素β4亞單位基因的突變和編碼18OkDa的大皰性類天皰瘡抗原BPAG2,也稱Ⅶ型膠原基因的突變。營養不良型大皰性表皮鬆解症的組織松解發生在緻密帶下錨原纖維水平,目前只發現Ⅶ型膠原基因(COL7A1)突變。

(二)發病機制
疾病的分子病理生理學 ;角蛋白多肽的突變位點與單純性大皰性表皮鬆解症的嚴重性之間有密切關係。D-M型角蛋白突變位於多肽中央螺旋桿區的氨基(1A)或羥基(2B)端,K型突變的位置較傾向於桿區的中央部分,w-c型突變位置經常或者位於桿區的非螺旋連接(L12)區,或位K5的前端。
1.單純型大皰性表皮鬆解症(Epidermolysis bullosa simplex,EBS)遺傳學基礎 單純型大皰性表皮鬆解症患者角蛋白K5及K14基因的分析,發現了角蛋白三種主要亞型的突變。功能研究顯示這些突變導致了,疾病。此病基因定位於染色體12qll~q13或17q12~q21,角蛋白K5和K14分別位於兩個位點。因此,單純型大皰性表皮鬆解症是因特異性基本角蛋白基因缺陷引起。已報導的大多數病例存在這兩角蛋白基因編碼區的點突變。然而,基因缺陷也可能位於K5和K14基因以外。最近發現,伴有肌營養小良的單純性大皰性表皮鬆解症與一種角蛋白絲相關蛋白(plectin)突變相關。因為角蛋白基因及轉錄產物長度(1.8~2.1kDa)較小,單純型大皰性表皮鬆解症患者角蛋白突變的篩查大多由DNA測序來實施。特別是當可進行皮膚活檢.角質形成細胞培養及mRNA提取時。如果一種抗體被用於診斷和分析,那麼針對角蛋白多肽關鍵區域的一組抗體的產生可有利於將來的診斷。此外,隨著形態-敏感膠電泳(CSGE)等方法的引入,可對DNA單個鹼基的改變做出快速檢測。對角蛋白基因突變的篩查現存也可變得更加簡易了。此方法在涉及大量患者標本篩查時尤其有用。它也消除了對整條基岡或轉錄基因測序的需要。
2.營養不良型大皰性表皮鬆解症(dystrophic)遺傳學基礎 在正常皮膚,Ⅶ型膠原分了形成反向二聚體,通過重疊的羧基末端連接起來。這種聯結通過鏈內的二硫鍵加強。這種穩固的Ⅶ型膠原分子側向聚集形成錨原纖維。這樣,Ⅶ型膠原合成後,進一步裝配成錨原纖維。因此,在轉錄或翻譯水平影響Ⅶ型膠原合成或干擾其超分子裝配成錨原纖維的突變都可表現為營養不良型大皰性表皮鬆解症。
對於HS-RDEB,目前發現患者Ⅶ型膠原的兩個等位基因的提前終止密碼子(PTC)的突變基因有低水平表達,但翻譯的蛋白在其羧基末端被截斷,不能裝配成錨原纖維。這種與HS-RIDEB超微結構中完全缺乏錨原纖維的改變一致,這也可解釋此型的特點皮膚極度脆弱。在輕型RDEB,等位基因可以編碼全長的Ⅶ型膠原多肽,但常發生錯義突變從而改變了蛋白質的空間構象,因此影響了錨原纖維裝配。
目前檢測到顯性遺傳型大皰性表皮鬆解症的突變是發生在膠原分子內以重複Gly-X-Y胺基酸序列為特徵的結構域的甘氨酸殘基替代。甘氨酸替代使膠原三環結構不穩定,干擾了其分泌,並且使其易於在胞外降解。因此,甘氨酸替代的作用是在翻譯後水平。由於Ⅶ型膠原是由三個相同的α1(Ⅶ)多肽組成的同質二聚體,則1/8的三環分子是正常的。因此口可形成一些正常的錨原纖維,這與超微結構觀察到的細錨原纖維和DDEB相對較輕的臨床表現一致。除了經典的DDEB型外,兩種臨床亞型(脛前營養不良型大皰性表皮鬆解症和Bart症候群)中存在甘氨酸替代突變。
3.交界型大皰性表皮鬆解症(JEB)遺傳學基礎 與前兩型大皰性表皮鬆解症中觀察到的基因純合性不同,交界型大皰性表皮鬆解症顯示很高程度的基因雜合性,目前認為至少六個不同的基因與其發病有關。在交界型大皰性表皮鬆解症(JEB),水皰發生在真皮表皮交界的基底膜內,即透明帶或重疊的半橋粒水平。電鏡下,觀察到半橋粒錨絲複合體區有異常。對大量致死性和非致性交界型大皰性表皮鬆解症患者的研究發現,編碼錨細絲蛋白-層粘連蛋白5的3個組成多肽α3、β3和β2的3個基因發生特異突變。最近,在交界型大皰性表皮鬆解症的一些亞型中檢測到編碼半橋粒其他成分的基因發生突變。例如,在一個大皰性表皮鬆解症合併幽門閉鎖患者身上檢測到編碼表皮細胞特異性整合素α6、β4的亞單位β4的基因突變。交界型大皰性表皮鬆解症中,臨床表現較輕的全身性營養不良性良性大皰性表皮鬆解症的患者顯示編碼180kDa的大皰性類天皰瘡抗原2(BPAG2,亦稱為XⅦ型膠原)的基因突變。日前對交界型大皰性表皮鬆解症分子基礎的認識強調半橋粒一錨細絲複合體的複雜性和在發病中的作用。
Herlitz型交界型大皰性表皮鬆解症,突變檢測顯示層粘連蛋白5每個基因均有突變(LAMA3、LAMB3和LAMC3,3個基因分別編碼α3、β3和γ2鏈)。發現大多數突變發生於LAMB3基因,存在著導致突變的兩個熱點區,即R42x和R635x。同時目前發現的所有突變均導致提前終止密碼子的產生,從而通過反義介導的mRNA降解機制使相應的mRNA轉錄降至很低水平。非Herlitz型交界型大皰性表皮鬆解症也有層粘連蛋白5基因的突變。在某些病例,層粘連蛋白5基因的其中一個基因的突變是提前終止密碼。然而,其他的基因突變是錯義突變或框架內外顯子跳躍突變,在這些病例中發現兩個區域基因突變。這些研究顯示帶完整羧基末端的全長多肽能裝配成三維分子。三維結構分子在錨細絲中有一定作用。
有一些患者的大皰發生在半橋粒內及其周同並且依其組織的超微結構的分類和臨床病情嚴重程度被認為「假交界型」。半橋粒(HD)的主要組成成分主要是命名為HDl~HD5的多肽。這些多從的異常很可能是交界型大皰性表皮鬆解症這些亞型的原因。根據超微結構的變化,在半橋粒水平發牛水皰的大皰性表皮鬆解症患者至少可分為3類。其臨床表現不同於任何一類經典的大皰性表皮鬆解症。它們是全身營養不良性良性大皰性表皮鬆解症(GABEB)、伴幽門閉鎖的大皰性表皮鬆解症(PA-JEB)和伴肌營養不良性大皰性表皮鬆解症(EB/MD)。在非致死性大皰性表皮鬆解症的特殊亞型即全身性營養不良性良性大皰性表皮鬆解症中,發現BPAG2基因發生突變。非致死性交界型大皰性表皮鬆解症的另一罕見亞型的特點為幽門狹窄和皮膚起皰為其首發症狀,它是β整合素基岡的突變的結果。
手、足部厚壁水皰的診斷
1.單純型大皰性表皮鬆解症(EBS) 是以表皮內水皰為特徵,主要由角蛋白突變所引起的一組遺傳性皮膚病,侵襲1/4萬人群。根據臨床的嚴重性進一步分成不同亞型。單純型大皰性表皮鬆解症家族的外顯率高,且它最嚴重的亞型,疾病在出生時就表現明顯。
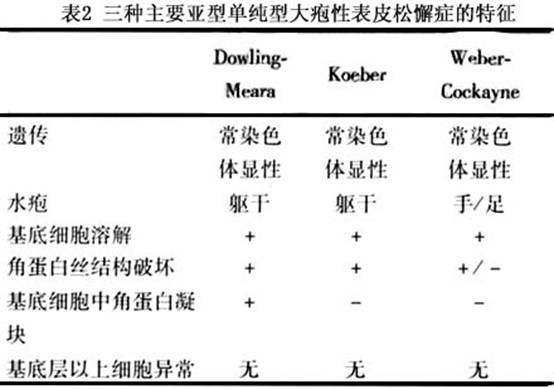
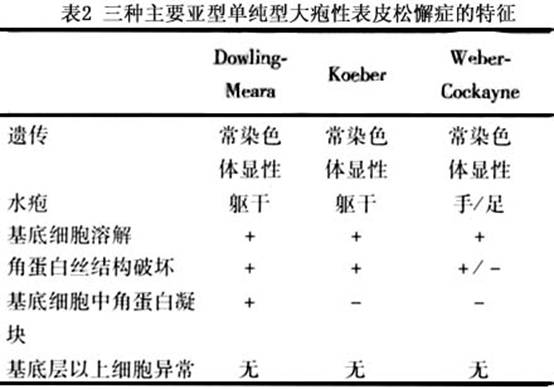
至少有11種亞型的單純型大皰性表皮鬆解症,其中7種為常染色體顯性遺傳。3種最常見亞型均為常染色體顯性遺傳,包括泛發性大皰性表皮鬆解症(Koebnet)、局限性大皰性表皮鬆解症(Weber Cockayne)和皰疹樣大皰性表皮鬆解症(Dowling Meata,表2)。隨著年齡增長起皰可顯著減少,有時可幾個月不起皰,可能是隨著患者年齡長大,表皮充分伸展,其所受的機械性張力自然減小。
(1)泛發性大皰性表皮鬆解症:始於新生兒至嬰兒早期,多見於手、足和四肢。也可見掌跖過度角化和脫屑。多不累及甲、齒和口腔黏膜。
(2)局限性大皰性表皮鬆解症:始於兒童時期或更晚,是最常見的一型。也可以到成人時才出現,表現為在高強度運動後出現手、足部厚壁水皰。常見手足多汗。足部的水皰常繼發感染。
(3)皰疹樣大皰性表皮鬆解症:出生時即可見,是最嚴重的一型,水皰泛發全身,可累及口腔黏膜。嬰兒期可出現明顯的炎症伴粟粒疹,在兒童早期水皰多不結痂。軀幹部和四肢近端可白髮成群或「皰疹樣」水皰,因為水皰裂隙位於表皮內,愈後不留瘢痕。指(趾)甲可能脫失,但通常可再生。
與前兩型不同,遇熱後水皰不會加重。到6、7歲時可出現掌跖角化過度。儘管一些患者水皰非常嚴重,但很少危及生命。因為局限性皮膚屏障功能喪失,易於繼發感染。
伴肌營養不良的單純型大皰性表皮鬆解症是惟一非角蛋白突變的單純型大皰性表皮鬆解症,與Koeber型類似,但成人期出現肌營養不良表現。
2.營養不良型大皰性表皮鬆解症 在水皰形成後癒合常伴有瘢痕和粟粒疹形成。因錨絲Ⅶ型膠原突變而致表皮下水皰。主要包括4種亞型,即Cockayne Touraine顯性遺傳型、Pasini白色丘疹樣顯性遺傳型、局限型隱性遺傳型和泛發性隱性遺傳型。此外,還有一些罕見亞型。如Bart症候群,新生兒暫時性大皰性表皮鬆解症等。
(1)顯性遺傳型:Cockayne Touraine。病中,水皰多見下肢端,有甲營養不良,始於嬰兒或兒童早期。癒合後因增生而形成瘢痕和粟粒疹。口腔損害不常見,牙齒多正常。Pasini型多始於出生時,水皰密集伴萎縮性瘢痕和粟粒疹。在無明顯外傷的情況下。膚色、瘢痕樣丘疹自發出現於軀下,稱為白色丘疹樣損害(albopapuloid lesions)。後期水皰主要局限於四肢,偶發全身。常見甲營養不良或甲缺失。黏膜表面和牙齒輕度累及。
(2)隱性遺傳型:臨床表現多樣。不嚴重的局限損害稱為mitis(輕)型,見於出生時,常累及肢端,伴關節表面萎縮性瘢痕和甲營養不良,但黏膜很少累及。表現較輕的局限性損害與局限性顯性遺傳型不易區別。嚴重型損害具致殘性,稱為Hallopeau-Siemens(HS-RDEB)型。出生時現廣泛的水皰,嬰兒期繼續擴展導致明顯的瘢痕形成。獲得性並指常導致手足部出現「拳擊手套」樣畸形。瘢痕從近端發展,進而累及整個肢體,形成彎曲攣縮。可累及甲、齒和頭皮。多數黏膜表面持續累及,伴複發性水皰和糜爛,導致食管狹窄和蹼化、尿道和肛門狹窄、包莖和角膜瘢痕。常合併營養不良、生長遲緩和慢性混合性貧血HS-RDEB最嚴重的合併症是在慢性糜爛區域發展為鱗狀細胞癌。高於50%的HS-RDEB患者在30歲左右時發展為此癌,許多死於癌轉移。
(3)Bart症候群:為DDEB的臨床亞型,呈常染色體顯性遺傳,是由Bart等首先報導,以先天性局限性皮膚缺損、機械性水皰和甲畸形為特徵的疾病,預後較好。
(4)新牛兒暫時性大皰性表皮鬆解症:1985年,Hashimoto等報導一新生兒在每次的輕微損傷後,皮膚上出現水皰、基底膜下發生分離、膠原和錨絲變性。至4個月大時很快痊癒。甲無任何損傷,皮損愈後無瘢痕形成。一般認為本病有以下特點:①出生時或摩擦誘發水皰、大皰性皮疹。②出上幾個月後可自行恢復。③無營養不良性瘢痕。④表皮下皰始於真皮乳頭層。⑤電鏡觀察到膠原溶解和破壞的錨絲。⑥粗內質網內有角質形成細胞的星狀體。
3.交界型大皰性表皮鬆解症(JEB) 至少存在6種臨床、亞型,最常見的有3型,Herlitz型、mitis型和泛發性良性營養不良型(GABEB)。
(1)Herlitz型:又稱為致死型和gravis型,常不能存活過嬰兒期,40%多在出生後l年內夭折。是所有大皰性表皮鬆解症中最嚴重的一型。出生時可見到泛發性水皰,伴嚴重的口周肉芽組織。甲常在早期脫失,再生時表現為甲營養不良。齒因釉質缺失而營養不良,多數黏膜表面有慢性侵蝕。頭皮損害常可見到慢性不癒合的侵蝕伴增殖性肉芽組織。系統損害包括整個上皮水皰,伴呼吸、胃腸道和生殖泌尿系統累及。常合併有氣管水皰、狹窄或阻塞,聲音嘶啞是早期嬰兒惡化的徵兆。顯著的生長遲緩和頑固性混合性貧血使治療更加困難。患兒常死於敗血症、多器官衰竭和營養不良。少見的臨床表現包括幽門和十二指腸閉鎖,因整合素基因突變而皮膚黏膜脆性極高。幽門閉鎖多合併有泌尿系統異常如腎盂積水和腎炎。
(2)Mitis(輕)型:又稱為非致死型,一些患兒在出生時表現為中等度的交界性損害,或雖為嚴重性損害但可存活過嬰兒期,並隨年齡增長而緩解。聲音嘶啞多較輕或無。頭皮與甲損害較明顯,口周不癒合的損害多見於4~10歲的患兒。罕見表現包括四肢或皺褶部位出現交界性水皰。
(3)GABEB:為非致死型的亞型,出生時出現全身皮膚累及。主要在四肢出現大小不一的水皰,軀幹、頭皮和面部也可累及。可持續至成人,伴有四肢、軀幹和頭皮部的漿液性或血性水皰和慢性損害。在溫度升高時水皰增多增大。水皰萎縮性癒合是本型的特有表現。甲可出現嚴重的營養不良。常見有或無瘢痕性脫髮。可有輕度口腔黏膜累及及因釉質缺失而致齒營養不良。水皰隨年齡增長而改善,但牙齒異常和萎縮瘢痕損害可持續至成人。生長正常,貧血罕見。
1.為了正確診斷單純型大皰性表皮鬆解症,需做皮膚活檢。皮膚活檢的超微結構分析可以明確皮膚中裂隙的位置,因此將單純型大皰性表皮鬆解症同其他類型的EB區別開來。它也可進一步區別其他水皰性皮膚病,例如表皮鬆解性角化過度(EH),它在病理上與單純型大皰性表皮鬆解症相似,但侵犯表皮基底細胞上層而不是基底細胞層。在少數病例中,此方法也被用做產前診斷。現在對單純型大皰性表皮鬆解症遺傳學基礎的理解使產前遺傳學諮詢成為可能,它可在孕早期進行,對胎兒的危險比皮膚活檢小。
2.突變分析在大皰性表皮鬆解症產前診斷中的應用 就遺傳諮詢、依據DNA的產前診斷和基因治療而言,準確了解不同亞型營養不良型大皰表皮鬆解症的致病突變基因可用來解釋一些問題。
(1)與患者關係較直接的是DNA的產前診斷,其最早可在妊娠10周時通過絨毛膜取樣檢查,或者在12~15周時經腹壁羊膜穿刺術檢查。對嚴重的營養不良型大皰性表皮鬆解症,可通過直接突變分析或遺傳連鎖分析進行產前DNA診斷。目前還沒有發現證明基因雜合性的其他方法。以上辦法已被用於有發生嚴重的致殘性RDEB的30多家庭的DNA產前診斷。這些遺傳學知識也會為發展通過分裂球分析進行植入前診斷提供基礎,這個技術進步可避免發現患病胎兒時必須終止妊娠。
(2)診斷和遺傳諮詢:營養不良型大皰性表皮鬆解症可以常染色體顯性和常染色體隱性遺傳的方式遺傳。對有嚴重的致殘性瘢痕的典型的HS-RDEB患者的診斷,即使臨床上其父母未發病,通常也不難診斷。同樣,垂直遺傳起皰趨向和相對輕的瘢痕表現型,在幾代人中有多位家庭成員受累,此時診斷顯性遺傳營養不良型大皰性表皮鬆解症無疑。
臨床上,在父母正常而患者表現較輕的診斷和確定遺傳類型時比較困難。超微結構觀察發現這些患者的皮膚雖有錨原纖維,但量少。常診斷為顯性營養不良型大皰性表皮鬆解症。認為其乃一新的顯性突變或親代種系嵌合。就對患者個體的基岡諮詢而言,這個診斷顯然有重要意義。若他們的疾病真是一新顯性突變,那麼其後代的發病率是1/2。相反,隱性遺傳性疾病其後代受累的危險大約和普通人群一樣低,但除外近親婚配。
對表現較輕,超微結構檢測到鋪原纖維,Ⅶ型膠原免疫熒光染色陽性的幾例患者基因突變和表現型的仔細鑒定提示其中許多是複合雜合子或隱性遺傳的純合性錯義突變。例如,第1個證叫輕型營養不良型大皰性表皮鬆解症是Ⅶ型膠原異常的事實揭示了純合性錯義突變,即在分子的羧基末端賴氨酸代替蛋氨酸(M2798K)。同樣,其他病例,1個等位基因的錯義突變,包括在膠原分子結構域的H-氨酸替代和另外一等位基因提前終止密碼的突變,可引起輕型RDEB。最後,對100多家庭的調查發現顯著的COL7Al突變,只有幾例顯示de noco顯性突變,而且至少有一個是來自其母系。基於上述考慮,在遺傳諮詢時,將每一「新」病例考慮為隱性遺傳較恰當,除非通過分子遺傳分析證明是顯性突變。依致病突變基因對營養不良型大皰性表皮鬆解症重新分類顯然利於估計患病個體的子代受累的可能性大小。
(3)準確理解引起交界型大皰性表皮鬆解症的突變在遺傳病諮詢、DNA為基礎的產前診斷和基因治療方面有意義。產前診斷最早可在妊娠10周時通過絨毛膜取樣檢查,或者在12~15周時經腹壁羊膜穿刺術檢查。因為引起交界型大皰性表皮鬆解症基因雜合性的組合很多,並且由於至少7個不同的基因可引起不同類型的交界型大皰性表皮鬆解症的基因變化,同時又觀察到熱點突變更新,因而產前診斷必須依據發現兩個突變的缺失或存在的直接證據。這些方法已被用於對許多有發生Herlitz型交界型大皰性表皮鬆解症危險性家庭的DNA產前診斷。
(4)中醫病機和辨證 中醫認為本病多因先天虧損,胎元不足,稟賦不充,脾腎陽虛;或因稟受胞中遺濕、遺熱、遺毒,復受外界摩擦而發病。
中醫辨證分型:
①脾虛濕盛型 一般健康狀況尚可,水皰大小不等,緊張豐滿,內容為漿液性,周圍無炎症,便溏。舌質淡、體胖有齒痕,苔白或白膩,脈沉緩。
辨證:脾虛濕盛、水濕外溢。
②脾腎陽虛型 多見嬰兒及兒童期,患兒身體瘦弱,頭髮稀疏,細軟或有脫髮,牙齒髮育不良,指甲軟或脫落,手足不溫,或常發青紫,常有五更瀉,皮膚有大皰或小皰,舌質淡或舌體胖嫩,苔白或少,脈沉細。此型多見於顯性營養不良型。
手、足部厚壁水皰的鑒別診斷
1.新生兒膿皰瘡 極易傳染,可呈流行性。
水皰易破裂,內容迅速變為膿性,可查見葡萄球菌或鏈球菌,炎症顯著,易於治癒。
2.皮膚卟啉病 水皰多見於於背、面部、耳等曝光部位,對光敏感。可見多毛,常伴發肝損害。尿及糞中尿卟啉及糞卟啉增高。
3.兒童線狀IgA大皰性皮病 發病不限於摩擦部位,無遺傳史,愈後不留萎縮性瘢痕。直接免疫熒光檢查可見IgA沿基底膜帶呈線狀沉積。
4.新生兒天皰瘡 往往泛發全身,皰壁鬆弛,用抗生素可迅速控制。
5.大皰性丘疹性蕁麻疹 常伴有明顯的瘙癢,且有水腫性丘疹存在。
6.獲得性大皰性表皮鬆解症 可由藥物、感染、卟啉病,澱粉樣變等引起,常伴有相關疾病的其他表現。
此外,在青春期,足部的水皰應與足癬、卟啉病等相鑒別。
1.單純型大皰性表皮鬆解症(EBS) 是以表皮內水皰為特徵,主要由角蛋白突變所引起的一組遺傳性皮膚病,侵襲1/4萬人群。根據臨床的嚴重性進一步分成不同亞型。單純型大皰性表皮鬆解症家族的外顯率高,且它最嚴重的亞型,疾病在出生時就表現明顯。
至少有11種亞型的單純型大皰性表皮鬆解症,其中7種為常染色體顯性遺傳。3種最常見亞型均為常染色體顯性遺傳,包括泛發性大皰性表皮鬆解症(Koebnet)、局限性大皰性表皮鬆解症(Weber Cockayne)和皰疹樣大皰性表皮鬆解症(Dowling Meata,表2)。隨著年齡增長起皰可顯著減少,有時可幾個月不起皰,可能是隨著患者年齡長大,表皮充分伸展,其所受的機械性張力自然減小。
(1)泛發性大皰性表皮鬆解症:始於新生兒至嬰兒早期,多見於手、足和四肢。也可見掌跖過度角化和脫屑。多不累及甲、齒和口腔黏膜。
(2)局限性大皰性表皮鬆解症:始於兒童時期或更晚,是最常見的一型。也可以到成人時才出現,表現為在高強度運動後出現手、足部厚壁水皰。常見手足多汗。足部的水皰常繼發感染。
(3)皰疹樣大皰性表皮鬆解症:出生時即可見,是最嚴重的一型,水皰泛發全身,可累及口腔黏膜。嬰兒期可出現明顯的炎症伴粟粒疹,在兒童早期水皰多不結痂。軀幹部和四肢近端可白髮成群或「皰疹樣」水皰,因為水皰裂隙位於表皮內,愈後不留瘢痕。指(趾)甲可能脫失,但通常可再生。
與前兩型不同,遇熱後水皰不會加重。到6、7歲時可出現掌跖角化過度。儘管一些患者水皰非常嚴重,但很少危及生命。因為局限性皮膚屏障功能喪失,易於繼發感染。
伴肌營養不良的單純型大皰性表皮鬆解症是惟一非角蛋白突變的單純型大皰性表皮鬆解症,與Koeber型類似,但成人期出現肌營養不良表現。
2.營養不良型大皰性表皮鬆解症 在水皰形成後癒合常伴有瘢痕和粟粒疹形成。因錨絲Ⅶ型膠原突變而致表皮下水皰。主要包括4種亞型,即Cockayne Touraine顯性遺傳型、Pasini白色丘疹樣顯性遺傳型、局限型隱性遺傳型和泛發性隱性遺傳型。此外,還有一些罕見亞型。如Bart症候群,新生兒暫時性大皰性表皮鬆解症等。
(1)顯性遺傳型:Cockayne Touraine。病中,水皰多見下肢端,有甲營養不良,始於嬰兒或兒童早期。癒合後因增生而形成瘢痕和粟粒疹。口腔損害不常見,牙齒多正常。Pasini型多始於出生時,水皰密集伴萎縮性瘢痕和粟粒疹。在無明顯外傷的情況下。膚色、瘢痕樣丘疹自發出現於軀下,稱為白色丘疹樣損害(albopapuloid lesions)。後期水皰主要局限於四肢,偶發全身。常見甲營養不良或甲缺失。黏膜表面和牙齒輕度累及。
(2)隱性遺傳型:臨床表現多樣。不嚴重的局限損害稱為mitis(輕)型,見於出生時,常累及肢端,伴關節表面萎縮性瘢痕和甲營養不良,但黏膜很少累及。表現較輕的局限性損害與局限性顯性遺傳型不易區別。嚴重型損害具致殘性,稱為Hallopeau-Siemens(HS-RDEB)型。出生時現廣泛的水皰,嬰兒期繼續擴展導致明顯的瘢痕形成。獲得性並指常導致手足部出現「拳擊手套」樣畸形。瘢痕從近端發展,進而累及整個肢體,形成彎曲攣縮。可累及甲、齒和頭皮。多數黏膜表面持續累及,伴複發性水皰和糜爛,導致食管狹窄和蹼化、尿道和肛門狹窄、包莖和角膜瘢痕。常合併營養不良、生長遲緩和慢性混合性貧血HS-RDEB最嚴重的合併症是在慢性糜爛區域發展為鱗狀細胞癌。高於50%的HS-RDEB患者在30歲左右時發展為此癌,許多死於癌轉移。
(3)Bart症候群:為DDEB的臨床亞型,呈常染色體顯性遺傳,是由Bart等首先報導,以先天性局限性皮膚缺損、機械性水皰和甲畸形為特徵的疾病,預後較好。
(4)新牛兒暫時性大皰性表皮鬆解症:1985年,Hashimoto等報導一新生兒在每次的輕微損傷後,皮膚上出現水皰、基底膜下發生分離、膠原和錨絲變性。至4個月大時很快痊癒。甲無任何損傷,皮損愈後無瘢痕形成。一般認為本病有以下特點:①出生時或摩擦誘發水皰、大皰性皮疹。②出上幾個月後可自行恢復。③無營養不良性瘢痕。④表皮下皰始於真皮乳頭層。⑤電鏡觀察到膠原溶解和破壞的錨絲。⑥粗內質網內有角質形成細胞的星狀體。
3.交界型大皰性表皮鬆解症(JEB) 至少存在6種臨床、亞型,最常見的有3型,Herlitz型、mitis型和泛發性良性營養不良型(GABEB)。
(1)Herlitz型:又稱為致死型和gravis型,常不能存活過嬰兒期,40%多在出生後l年內夭折。是所有大皰性表皮鬆解症中最嚴重的一型。出生時可見到泛發性水皰,伴嚴重的口周肉芽組織。甲常在早期脫失,再生時表現為甲營養不良。齒因釉質缺失而營養不良,多數黏膜表面有慢性侵蝕。頭皮損害常可見到慢性不癒合的侵蝕伴增殖性肉芽組織。系統損害包括整個上皮水皰,伴呼吸、胃腸道和生殖泌尿系統累及。常合併有氣管水皰、狹窄或阻塞,聲音嘶啞是早期嬰兒惡化的徵兆。顯著的生長遲緩和頑固性混合性貧血使治療更加困難。患兒常死於敗血症、多器官衰竭和營養不良。少見的臨床表現包括幽門和十二指腸閉鎖,因整合素基因突變而皮膚黏膜脆性極高。幽門閉鎖多合併有泌尿系統異常如腎盂積水和腎炎。
(2)Mitis(輕)型:又稱為非致死型,一些患兒在出生時表現為中等度的交界性損害,或雖為嚴重性損害但可存活過嬰兒期,並隨年齡增長而緩解。聲音嘶啞多較輕或無。頭皮與甲損害較明顯,口周不癒合的損害多見於4~10歲的患兒。罕見表現包括四肢或皺褶部位出現交界性水皰。
(3)GABEB:為非致死型的亞型,出生時出現全身皮膚累及。主要在四肢出現大小不一的水皰,軀幹、頭皮和面部也可累及。可持續至成人,伴有四肢、軀幹和頭皮部的漿液性或血性水皰和慢性損害。在溫度升高時水皰增多增大。水皰萎縮性癒合是本型的特有表現。甲可出現嚴重的營養不良。常見有或無瘢痕性脫髮。可有輕度口腔黏膜累及及因釉質缺失而致齒營養不良。水皰隨年齡增長而改善,但牙齒異常和萎縮瘢痕損害可持續至成人。生長正常,貧血罕見。
1.為了正確診斷單純型大皰性表皮鬆解症,需做皮膚活檢。皮膚活檢的超微結構分析可以明確皮膚中裂隙的位置,因此將單純型大皰性表皮鬆解症同其他類型的EB區別開來。它也可進一步區別其他水皰性皮膚病,例如表皮鬆解性角化過度(EH),它在病理上與單純型大皰性表皮鬆解症相似,但侵犯表皮基底細胞上層而不是基底細胞層。在少數病例中,此方法也被用做產前診斷。現在對單純型大皰性表皮鬆解症遺傳學基礎的理解使產前遺傳學諮詢成為可能,它可在孕早期進行,對胎兒的危險比皮膚活檢小。
2.突變分析在大皰性表皮鬆解症產前診斷中的應用 就遺傳諮詢、依據DNA的產前診斷和基因治療而言,準確了解不同亞型營養不良型大皰表皮鬆解症的致病突變基因可用來解釋一些問題。
(1)與患者關係較直接的是DNA的產前診斷,其最早可在妊娠10周時通過絨毛膜取樣檢查,或者在12~15周時經腹壁羊膜穿刺術檢查。對嚴重的營養不良型大皰性表皮鬆解症,可通過直接突變分析或遺傳連鎖分析進行產前DNA診斷。目前還沒有發現證明基因雜合性的其他方法。以上辦法已被用於有發生嚴重的致殘性RDEB的30多家庭的DNA產前診斷。這些遺傳學知識也會為發展通過分裂球分析進行植入前診斷提供基礎,這個技術進步可避免發現患病胎兒時必須終止妊娠。
(2)診斷和遺傳諮詢:營養不良型大皰性表皮鬆解症可以常染色體顯性和常染色體隱性遺傳的方式遺傳。對有嚴重的致殘性瘢痕的典型的HS-RDEB患者的診斷,即使臨床上其父母未發病,通常也不難診斷。同樣,垂直遺傳起皰趨向和相對輕的瘢痕表現型,在幾代人中有多位家庭成員受累,此時診斷顯性遺傳營養不良型大皰性表皮鬆解症無疑。
臨床上,在父母正常而患者表現較輕的診斷和確定遺傳類型時比較困難。超微結構觀察發現這些患者的皮膚雖有錨原纖維,但量少。常診斷為顯性營養不良型大皰性表皮鬆解症。認為其乃一新的顯性突變或親代種系嵌合。就對患者個體的基岡諮詢而言,這個診斷顯然有重要意義。若他們的疾病真是一新顯性突變,那麼其後代的發病率是1/2。相反,隱性遺傳性疾病其後代受累的危險大約和普通人群一樣低,但除外近親婚配。
對表現較輕,超微結構檢測到鋪原纖維,Ⅶ型膠原免疫熒光染色陽性的幾例患者基因突變和表現型的仔細鑒定提示其中許多是複合雜合子或隱性遺傳的純合性錯義突變。例如,第1個證叫輕型營養不良型大皰性表皮鬆解症是Ⅶ型膠原異常的事實揭示了純合性錯義突變,即在分子的羧基末端賴氨酸代替蛋氨酸(M2798K)。同樣,其他病例,1個等位基因的錯義突變,包括在膠原分子結構域的H-氨酸替代和另外一等位基因提前終止密碼的突變,可引起輕型RDEB。最後,對100多家庭的調查發現顯著的COL7Al突變,只有幾例顯示de noco顯性突變,而且至少有一個是來自其母系。基於上述考慮,在遺傳諮詢時,將每一「新」病例考慮為隱性遺傳較恰當,除非通過分子遺傳分析證明是顯性突變。依致病突變基因對營養不良型大皰性表皮鬆解症重新分類顯然利於估計患病個體的子代受累的可能性大小。
(3)準確理解引起交界型大皰性表皮鬆解症的突變在遺傳病諮詢、DNA為基礎的產前診斷和基因治療方面有意義。產前診斷最早可在妊娠10周時通過絨毛膜取樣檢查,或者在12~15周時經腹壁羊膜穿刺術檢查。因為引起交界型大皰性表皮鬆解症基因雜合性的組合很多,並且由於至少7個不同的基因可引起不同類型的交界型大皰性表皮鬆解症的基因變化,同時又觀察到熱點突變更新,因而產前診斷必須依據發現兩個突變的缺失或存在的直接證據。這些方法已被用於對許多有發生Herlitz型交界型大皰性表皮鬆解症危險性家庭的DNA產前診斷。
(4)中醫病機和辨證 中醫認為本病多因先天虧損,胎元不足,稟賦不充,脾腎陽虛;或因稟受胞中遺濕、遺熱、遺毒,復受外界摩擦而發病。
中醫辨證分型:
①脾虛濕盛型 一般健康狀況尚可,水皰大小不等,緊張豐滿,內容為漿液性,周圍無炎症,便溏。舌質淡、體胖有齒痕,苔白或白膩,脈沉緩。
辨證:脾虛濕盛、水濕外溢。
②脾腎陽虛型 多見嬰兒及兒童期,患兒身體瘦弱,頭髮稀疏,細軟或有脫髮,牙齒髮育不良,指甲軟或脫落,手足不溫,或常發青紫,常有五更瀉,皮膚有大皰或小皰,舌質淡或舌體胖嫩,苔白或少,脈沉細。此型多見於顯性營養不良型。
手、足部厚壁水皰的治療和預防方法
根據病因進行針對治療和預防。
參看
| 關於「手、足部厚壁水皰」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |