病理生理學/低鉀血症
| 醫學電子書 >> 《病理生理學》 >> 水和電解質代謝紊亂 >> 鉀代謝紊亂 >> 低鉀血症 |
| 病理生理學 |
|
|
血清鉀濃度低於3.5mmol/L(3.5mEq/L,正常人血清鉀濃度的範圍為3.5~5.5mmol/L)稱為低鉀血症。低鉀血症時,機體的含鉀總量不一定減少,細胞外鉀向細胞內轉移時,情況就是如此。但是,在大多數情況下,低鉀血症的患者也伴有體鉀總量的減少——缺鉀(potassium deficit)。
(一)原因和機制
1.鉀攝入減少一般飲食含鉀都比較豐富。故只要能正常進食,機體就不致缺鉀。消化道梗阻、昏迷、手術後較長時間禁食的患者,不能進食。如果給這些患者靜脈內輸入營養時沒有同時補鉀或補鉀不夠,就可導致缺鉀和低鉀血症。然而,如果攝入不足是唯一原因,則在一定時間內缺鉀程度可以因為腎的保鉀功能而不十分嚴重。當鉀攝入不足時,在4~7天內可將尿鉀排泄量減少到20mmol/L以下,在7~10天內則可降至5~10mmol/L(正常時尿鉀排泄量為38~150mmol/L)。
2.鉀排出過多
⑴經胃腸道失鉀:這是小兒失鉀最重要的原因,常見於嚴重腹瀉嘔吐等伴有大量消化液喪失的患者。腹瀉時糞便中K+的濃度可達30~50mmol/L。此時隨糞丟失的鉀可比正常時多10~20倍。糞鉀含量之所以增多,一方面是因為腹瀉而使鉀在小腸的吸收減少,另一方面是由於腹瀉所致的血容量減少可使醛固酮分泌增多,而醛固酮不僅可使尿鉀排出增多,也可使結腸分泌鉀的作用加強。由於胃液含鉀量只有5~10mmol/L,故劇烈嘔吐時,胃液的喪失並非失鉀的主要原因,而大量的鉀是經腎隨尿喪失的,因為嘔吐所引起的代謝性鹼中毒可使腎排鉀增多(詳後文),嘔吐引起的血容量減少也可通過繼發性醛固酮增多而促進腎排鉀。
⑵經腎失鉀:這是成人失鉀最重要的原因。引起腎排鉀增多的常見因素有:
①利尿藥的長期連續使用或用量過多:例如,抑制近曲小管鈉、水重吸收的利尿藥(碳酸酐酶抑製藥乙醯唑胺),抑制髓袢升支粗段Cl-和Na+重吸收的利尿藥(速尿、利尿酸、噻嗪類等)都能使到達遠側腎小管的原尿流量增加,而此處的流量增加是促進腎小管鉀分泌增多的重要原因。上述利尿藥還能使到達遠曲小管的Na+量增多,從而通過Na+-K+交換加強而導致失鉀。許多利尿藥還有一個引起腎排鉀增多的共同機制:通過血容量的減少而導致醛固酮分泌增多。速尿、利尿酸、噻嗪類的作用在於抑制髓袢升支粗段對Cl-的重吸收從而也抑制了Na+的重吸收。所以,這些藥物的長期使用既可導致低鈉血症,又可導致低氯血症。已經證明,任何原因引起的低氯血症均可使腎排鉀增多。其可能機制之一是低氯血症似能直接剌激遠側腎小管的泌鉀功能。
②某些腎臟疾病:如遠側腎小管性酸中毒時,由於遠曲小管泌氫功能障礙,因而H+-Na+交換減少而K+-Na+交換增多而導致失鉀。近側腎小管性酸中毒時,近曲小管HCO3-的重吸收減少,到達遠曲小管的HCO3-增多是促進遠曲小管排鉀增多的重要原因(詳後文)。急性腎小管壞死的多尿期,由於腎小管液中尿素增多所致的滲透性利尿,以及新生腎小管上皮對水、電解質重吸收的功能不足,故可發生排鉀增多。
③腎上腺皮質激素過多:原性和繼發懷醛固酮增多時,腎遠曲小管和集合管Na+-K+交換增加,因而起排鉀保鈉的作用。Cushing症候群時,糖皮質激素皮質醇的分泌大量增多。皮質醇也有一定的鹽皮質激素樣的作用。大量、長期的皮質醇增多也能促進遠曲小管和集合管的Na+-K+交換而導致腎排鉀增多。
④遠曲小管中不易重吸收的陰離子增多:HCO3-、SO42-、HPO42-、NO3-、β-羥丁酸、乙醯乙酸、青黴素等均屬此。它們在遠曲小管液中增多時,由於不能被重吸收而增大原尿的負電荷,因而K+易從腎小管上皮細胞進入管腔液而隨尿喪失。
⑤鎂缺失:鎂缺失常常引起低鉀血症。髓袢升支的鉀重吸收有賴於腎小管上皮細胞中的Na+-K+-ATR酶,而這種酶又需Mg2+的激活。缺鎂時,可能因為細胞內Mg2+缺失而使此酶失活,因而該處鉀重吸收發生障礙而致失鉀。動物實驗還證明,鎂缺失還可引起醛固酮增多,這也可能是導致失鉀的原因。
⑥鹼中毒:鹼中毒時,腎小管上皮細胞排H+減少,故H+-Na+交換加強,故隨尿排鉀增多。
(3)經皮膚失鉀:汗液含鉀只有9mmol/L。在一般情況下,出汗不致引起低鉀血症。但在高溫環境中進行重體力勞動時,大量出汗亦可導致鉀的喪失。
3.細胞外鉀向細胞內轉移 細胞外鉀向細胞內轉移時,可發生低鉀血症,但在機體的含鉀總量並不因而減少。
⑴低鉀性周期性麻痹:發作時細胞外鉀向細胞內轉移,是一種家族性疾病。
⑵鹼中毒:細胞內H+移至細胞外以起代償作用,同時細胞外K+進入細胞。
⑶過量胰島素:用大劑量胰島素治療糖尿病酮症酸中毒時,發生低鉀血症的機制有二:
①胰島素促進細胞糖原合成,糖原合成需要鉀,血漿鉀乃隨葡萄糖進入細胞以合成糖原。
②胰島素有可能直接剌激骨骼肌細胞膜上的Na+-K+-ATP酶,從而使肌細胞內Na+排出增多而細胞外K+進入肌細胞增多。
⑷鋇中毒:抗日戰爭時期四川某地發生大批「趴病」病例,臨床表現主要是肌肉軟弱無力和癱瘓,嚴重者常因呼吸肌麻痹而死亡。經我國學者杜公振等研究,確定該病的原因是鋇中毒。但當時鋇中毒引起癱瘓的機制尚未闡明。現已確證,鋇中毒引起癱瘓的機制在於鋇中毒引起了低鉀血症。鋇中毒時,細胞膜上的Na+-K+-ATP酶繼續活動。故細胞外液中的鉀不斷進入細胞。但鉀從細胞內流出的孔道卻被特異地阻斷,因而發生低鉀血症。引起鋇中毒的是一些溶於酸的鋇鹽如醋酸鋇、碳酸鋇、氯化鋇、氫氧化鋇、硝酸鋇和硫化鋇等。
4.粗製生棉油中毒 近二三十年來,在我國某些棉產區出現一種低血鉀麻痹症,在一些省內又被稱為「軟病」。其臨床主要特徵是四肢肌肉極度軟弱或發生弛緩性麻痹,嚴重者常因呼吸肌麻痹而死亡,血清鉀濃度明顯降低。往往在同一地區有許多人發病。病因與食用粗製生棉籽油有密切關係。粗製生棉油是農村一些小型油廠和榨坊生產的。這些廠的生產工藝不合規格。棉籽未經充分蒸炒甚至未曾脫殼就用來榨油,榨出的油又未按規定進行加鹼精鍊。因此棉籽中的許多毒性物質存於油中。與「軟病」的發生和隨後的一系列研究,都是棉酚(gossypol)。「軟病」時低鉀血症的發生機制尚未闡明。「軟病」的發現和隨後的一系列研究,都是我國學者進行的。迄今為止,國外的書刊中,尚無該病的記載。
(二)對機體的影響
低鉀血症對機體的影響,在不同的個體有很大的差別。低鉀血症的臨床表現也常被原發病和鈉水代謝紊亂所掩蓋。低鉀血症的症状取決於失鉀的快慢和血鉀降低的程度。失鉀快則症状出現快,而且也較嚴重;失鉀慢則缺鉀雖已較重,症状也不一定顯著。一般說來,血清鉀濃度愈低,症状愈嚴重。但有一點應當強調指出,在可興奮的組織內,興奮性不僅與血清鉀降低的程度有關。而更重要的還取決於細胞內鉀濃度與細胞外鉀濃度之比([K+]i/[K+]e)。比值大則興奮性減低,比值小則興奮性增高。
雖然細胞內的許多酶需要鉀激活,但是細胞內鉀濃度的輕度降低(例如從160降至130mmol/L)是否會明顯地影響這些酶的活性,尚不清楚。
動物實驗證明,缺鉀時細胞內外發生離子交換。即細胞內K+逸出而細胞外Na+和H+進入細胞。缺鉀比較嚴重時,細胞內Na+和H+的積聚可達到足以影響酶活性的程度。因此,缺鉀引起的細胞功能障礙很可能是細胞內鈉離子濃度和pH改變的結果。
低鉀血症對機體的影響如下:
1.對骨骼肌的影響 主要是超極化阻滯。低鉀血症時[K+]i/[K+]e的比值增大,因而肌細胞靜息電位負值增大。靜息電位與閾電位的距離增大,細胞興奮性於是降低,嚴重時甚至不能興奮,亦即細胞處於超極化阻滯狀態。臨床上先是出現肌肉無力。繼而可發生弛緩性麻痹。這種變化在四肢肌肉最為明顯,嚴重者可發生呼吸肌麻痹,這是低鉀血症患者的主要死亡原因之一(圖5-3)。
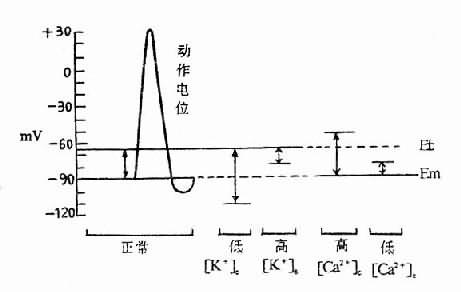
圖5-3 細胞外液K+、Ca2+濃度和正常骨骼肌靜息膜電位(Em)與閾電位(Et)的關係
肌肉興奮性的這種變化在急性缺鉀要比在慢性缺鉀時嚴重得多。因為在急性缺鉀時,細胞外鉀濃度已經顯著降低而細胞內鉀在短時間內尚來不及較多地外逸,故細胞內外鉀的濃度差明顯增大,[K+]i/[K+]e比值顯著增大。在慢性缺鉀時,隨著時間的推移。細胞內鉀釋出也較多,因而[K+]i/[K+]e比值變化可以不大。因此,同一水平的低鉀血症,在急性缺鉀患者可引起嚴重的肌肉麻痹而在慢性缺鉀患者卻可無明顯的肌肉症状。
2.對心臟的影響
⑴對興奮性的影響:按理論推測,細胞外液鉀濃度降低時,由於細胞膜內外K+濃度差增大,細胞內K+外流應當增多而使心肌細胞靜息電位負值增大而呈超極化狀態。但實際上當血清鉀濃度降低特別是明顯降低(如低於3mmol/L)時,靜息電位負值反而減少,這可能是由於細胞外液鉀濃度降低時,心肌細胞膜的鉀電導(potassium conductance)降低,從而使細胞內鉀外流減少,而基礎的內向鈉電流使膜部分去極化所致。靜息電位負值的減少使靜息電位與閾電位的距離減小,因而引起興奮所需的剌激也較小,所以心肌的興奮性增高。細胞外液鉀濃度降低時對鈣內流的抑制作用減小,故鈣內流加速而使復極化2期(坪期)縮短,心肌的有效不應期也隨之而縮短。心肌細胞膜的鉀電導降低所致的鉀外流減小,又使3期復極的時間延長。近年有人從低鉀血症病人的右心室尖部所記錄的心肌細胞動作電位中也觀察到3期復極時間的延長。3期復極時間的延長也就說明心肌超常期延長。上述變化使整個動作電位的時間延長,因而後一次0期除極化波可在前一次復極化完華之前到達。在心電圖上可見反映2期復極的S-T段壓低。相當於3期復極的T波壓低和增寬,並可在其末期出現明顯的U波,相當於心室動作電位時間的Q-T間期延長。
⑵對自律性的影響:在心房傳導組織、房室束-浦肯野纖維網的快反應自律細胞,當3期復極末達到最大復極電位(-90mV)後,由於膜上Ik通道通透性進行性衰減使細胞內鉀的外流逐漸減少,而鈉離子又從細胞外緩慢而不斷地進入細胞(背景電流),故進入細胞的正電荷量逐漸超過逸出細胞的正電荷量,膜就逐漸去極化,當到達閾電位時就發生0期去極化。這就是快反應細胞的自動去極化。在低鉀血症時鉀電導降低,故在到達最大復極電位後,細胞內鉀的外流比正常減慢而鈉內流相對加速。因而這些快反應自律細胞的自動去極化加速,自律性增高。
⑶對傳導性的影響:低鉀血症時因心肌靜息電位負值變小,去極化時鈉內流速度減慢。故0期膜內電位上升的速度減慢,幅度減小,興奮的擴布因而減慢,心肌傳導性降低。在心電圖上,可見P-R間期延長,說明去極化波由心房傳導到心室所需的時間延長,QRS綜合波增寬,說明心室內傳導性降低(圖5-4)。
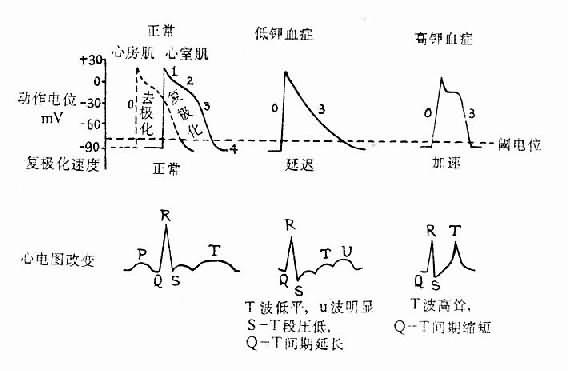
圖5-4 血漿鉀濃度對心肌細胞膜電位及心電圖的影響
由上述可見,低鉀血症時由於心肌興奮性增高、超常期延長和異位起搏點自律性增高等原因,容易發生心律失常。傳導性降低所致的傳導緩慢和單向傳導阻滯,加上有效不應期的縮短有助於興奮折返,因而也可引起包括心室纖維顫動在內的心律失常。
(4)對收縮性的影響:如前所述,細胞外液鉀濃度降低時對鈣內流的抑制作用減小,故在2期復極時鈣內流加速,心肌細胞內Ca2+濃度增高,興奮-收縮偶聯過程加強,心肌收縮性增強。然而,低鉀血症對心肌收縮性的影響因缺鉀的程度和持續時間而異:在早期或輕度低鉀血症時,心肌收縮性增強;但在嚴重的慢性缺鉀時,心肌收縮性減弱。與此相應的組織學變化是:在實驗動物的心肌中可見橫紋的消失、間質細胞浸潤、不同程度的心肌壞死和瘢痕形成。由此也可以理解,有些嚴重慢性缺鉀的狗,可因心力衰竭而發生肺水腫。然而在臨床上,缺鉀很少成為心力衰竭的原因。
3.對腎的影響
⑴尿濃縮功能障礙:在慢性缺鉀伴有低鉀血症時,常出現尿濃縮的障礙。由此可以理解,慢性缺鉀的病人常有多尿和低比重尿的臨床表現。尿濃縮功能障礙的發生機制在於:
①遠曲小管對ADH的反應性不足;
②低鉀血症時髓袢升支NaCL的重吸收不足以致髓質滲透壓梯度的形成發生障礙。
⑵腎血流量減少:人和動物缺鉀時都可發生腎血管收縮,從而引起腎血流量減少。引起腎血管收縮的因素有:
②血管緊張素Ⅱ的水平增高。
⑶腎小球濾過率減少:在實驗動物,腎小球濾過率的減少似與腎血流量的減少平行。在病人,嚴重而持續的缺鉀也可使腎小球濾過率明顯減少。時間久後,可導致腎的器質性損害。
⑷腎形態結構的變化:在大鼠,缺鉀引起的病變主要見於髓質集合管,表現為增殖性反應包括上皮細胞腫脹、增生和胞質內顯著的顆粒形成。持久的缺鉀可導致間質瘢痕形成、腎小球硬化和腎小管擴張等器質性變化。在人,慢性缺鉀主要引起近曲小管上皮細胞的空泡形成,也可發生間質瘢痕形成、間質淋巴細胞浸潤和腎小管萎縮等變化。
以上的變化中,除了顯著的纖維化和腎組織的喪失以外,一般都是可復性的。
4.對胃腸的影響
鉀缺乏可引起胃腸運動減弱。患者常發生噁心、嘔吐和厭食,嚴重缺鉀可致難以忍受的腹脹甚至麻痹性腸梗阻。
5.對代謝的影響
⑴糖代謝:血漿鉀濃度的降低可抑制胰腺分泌胰島素,因而低鉀血症患者的糖原合成發生障礙,對葡萄糖的耐量不足,易發生高血糖。應當看到,這時的胰島素分泌減少也有一定的代償意義,因為胰島素可通過保養進細胞內糖原合成和直接剌激骨骼肌細胞膜上的Na+K+-ATP酶而使細胞外鉀向組織內轉移。可見低鉀血症時的胰島素分泌減少,有助於防止血漿鉀濃度的進一步降低。
⑵蛋白代謝:缺鉀可以引起負氮平衡,因為鉀是蛋白合成所必需。在兒童,鉀缺乏可以成為生長障礙的原因之一。
⑶水、電解質和酸鹼平衡:
①醛固酮分泌減少;血漿鉀濃度降低能直接抑制腎上腺皮質球帶合成醛固酮。血漿醛固酮水平的降低能減少腎遠曲小管等對鉀的排泄,因而也有一定的代償意義;
②腎產氨增加:低鉀血症時可能通過細胞內酸中毒而使腎臟遠曲小管產氨增加,氨排出的增多可使遠曲小管排鉀減少,因而也有代償意義;
③多尿多飲:慢性缺鉀時,尿濃縮功能減退,因而排出大量低比重尿。水分的喪失引起渴感。動物實驗證明缺鉀也能剌激渴感,從而引起多飲。
④腎排氯增多:缺鉀時,全部腎小管特別是其遠側部分對氯的重吸收減少。
⑤酸鹼平衡:低鉀血症患者的酸鹼平衡狀態與原發疾病或引起低鉀血症的原因有密切關係。例如,當原發疾病為腎小管酸中毒,或引起缺鉀的原因為腹瀉時,患者就可伴有代謝性酸中毒。當引起缺鉀的原因是長時間應用高效能利尿藥如速尿、利尿酸時,患者就有代謝性鹼中毒。但是,缺鉀和低鉀血症本身卻往往傾向於引起代謝性鹼中毒。這是因為,第一,低鉀血症時,遠曲小管內K+-Na+交換減少,故H+-Na+交換增多,因而排H+增多;而且,如前所述,低鉀血症時腎遠曲小管產氨和排氨增多,氨又可與H+增多結合成NH4+而排出;第二,低鉀血症時(原因為細胞外鉀向細胞內轉移者除外),細胞內K+向細胞外釋出,細胞外的H+進入細胞,從而使細胞外液H+濃度降低;第三,如前所述,缺鉀時腎排氯增多,而機體缺氯可引起代謝性鹼中毒(參閱《酸鹼平衡紊亂》)。可見在一個具體的低鉀血症患者,酸鹼平衡的狀態是由原發疾病、缺鉀原因和低鉀血症本身的影響來共同決定的。
(三)防治原則
1.防治原發疾病,去除引起缺鉀的原因如停用某些利尿藥等。
2.補鉀如果低鉀血症較重(血清鉀低於2.5~3.0mmol/L)或者還有顯著的臨床表現如心律失常、肌肉癱瘓等,則應及時補鉀。
補鉀最好口服,每天以40~120mmol為宜。只有當情況危急,缺鉀即將引起威脅生命的併發症時,或者因噁心、嘔吐等原因使患者不能口服時才應靜脈內補鉀。而且,只有當每日尿量在500ml以上才容許靜脈內補鉀。輸入液的鉀濃度不得超過40mmol/L,每小時滴入的量一般不應超過10mmol。靜脈內補鉀時要定時測定血鉀濃度,作心電圖描記以進行監護。
細胞內缺鉀恢復較慢,有時需補鉀4~6日後細胞內外的鉀才能達到平衡,有的嚴重的慢性缺鉀患者需補鉀10~15日以上。
如低鉀血症伴有代謝性鹼中毒或酸鹼狀態無明顯變化,宜用KCL。KCL對各種原因引起的低鉀血症實際上也都適用,因為低鉀血症本身就可以引起缺氯。如低鉀血症伴有酸中毒,則可用KHCO3或檸檬酸鉀,以同時糾正低鉀血症和酸中毒。
3.糾正水和其它電解質代謝紊亂引起低鉀血症的原因中,有不少可以同時引起水和其他電解質如鈉、鎂等的喪失,因此應當及時檢查,一經發現就必須積極處理。如前所述,如果低鉀血症是由缺鎂引起,則如不補鎂,單純補鉀是無效的。
參看
| 關於「病理生理學/低鉀血症」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |