臨床生物化學/其它糖代謝異常
| 醫學電子書 >> 《臨床生物化學》 >> 糖代謝紊亂 >> 糖代謝的先天性異常 >> 其它糖代謝異常 |
| 臨床生物化學 |
|
|
|
6-磷酸葡萄糖脫氫酶(G-6-PD)催化6-磷酸葡萄糖脫氫生成6-磷酸葡萄內脂,脫下的氫由NADP接受。此步反應是磷酸戊糖途徑的關鍵部位,所產生的NADPH在維持紅細胞的正常形態與功能方面起了重要作用。
G-6-PD缺乏病是伴性遺傳性疾病,臨床並不罕見。女性雜合子含有兩族紅細胞:一族酶活性正常,另一族缺乏G-6-PD。輕型G-6-PD缺乏病人紅細胞中G-6-PD活性比正常人低10倍。在一般情況下磷酸戊糖途徑提供的NADPH還能維持還原型谷胱甘肽的水平,保證紅細胞的正常形態與功能。當紅細胞中NADPH的需要量增加,如服用抗瘧疾藥撲瘧喹啉時,正常人不會有什麼危害,而G-6-PD缺乏病人紅細胞中磷酸戊糖途徑的代謝速度則不能相應增加,提供的NADPH不能保證維持還原型谷胱甘肽所應有的水平,可引起嚴重的溶血性貧血,俗稱蠶豆黃。
(二)先天性家族性非溶血性黃疸
人類先天性家族性非溶血性黃疸(Grigler-Najjar症候群)是由於缺乏UDP-葡萄糖醛酸基轉移酶,使膽紅素不能與葡萄糖醛酸結合,形成結合膽紅素,使膽紅素以不易運輸和排泄的游離形在體內堆積所致的先天性疾病。
正常人通過糖醛酸途徑產生葡萄糖醛酸,後者在UDP-葡萄糖醛酸基轉移酶的催化下,可與內源性如類固醇、膽紅素和外源性如藥物等物質結合,生成相應的葡萄糖苷酸化合物。結合型的葡萄糖苷酸化合物具有較強的酸性,在生理pH下有較高的溶解度,易於運輸排泄。這在體內類固醇激素的滅活和膽紅素的代謝,以及許多生物轉化作用中具有重要意義。而病人因先天缺乏此酶,使其不但對膽紅素代謝造成異常,同時也缺乏結合外源性物質生成葡萄糖苷酸化合物的能力。
果糖是食物中糖的一部分,主要來自蔗糖。從腸道吸收的果糖大部分在肝內通過1-磷酸果糖(F-1-P)途徑代謝。代謝途徑見圖3-9。果糖代謝障礙是因與果糖代謝有關的酶缺乏所致。
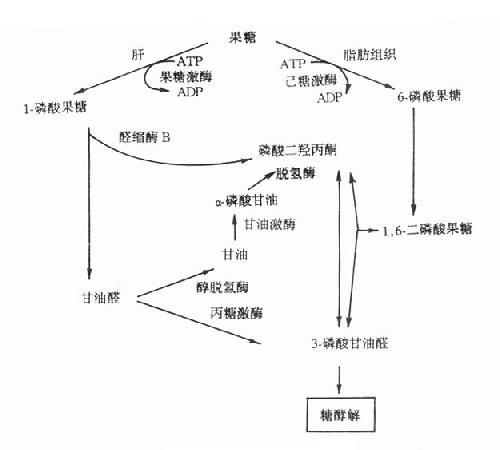
圖3-9 果糖代謝途徑
1.實質性果糖尿(essentialfructosuria)又稱為原發性果糖尿症,它是由於果糖激酶缺乏所引起的常染色體隱性遺傳疾病。正常人血中果糖比葡萄糖代謝快,其半壽期果糖為20分鐘,葡萄糖為45分鐘一次服用50g果糖後,通常在2小時之內血中果糖濃度就降至空腹水平(0-0.44mmol/L)。果糖激酶缺乏者一次服用50g果糖後,病人血中果糖濃度異常高,2小時內果糖仍未消失,並出現果糖尿。此型果糖尿又稱為Ⅰ型果糖尿。病人無低血糖表現,這是因為病人機體內葡萄糖與乳糖代謝均正常。
2.果糖不耐受(fructoseintolerance)此病為常染色體隱性遺傳性疾病,雜合子無症状。多數病人在斷奶後給予蔗糖飲食時才發病,嚴重病例可致死亡。有些病例可由於手術前後給予果糖或靜脈注射山梨醇引起嚴重肝、腎損傷時才發現。
果糖不耐受症的臨術表現可明顯不同。在嬰兒可表現為嘔吐、進食少、肝大、精神淡漠、生長停滯等。病人尿分析有果糖、葡萄糖等還原物質,多數病人有蛋白尿、非特異的胺基酸尿以及血漿轉酶活性增高。有的病兒伴有其它肝、腎損害指標。在大齡兒童或成人則無症状,只在進甜食後,可有腹部不適、嘔吐或腹瀉。果糖不耐受的一個重要特徵是服用果糖後出現嚴重的低血糖,病人即使肝糖原儲備豐富也會在此時發生低血糖,這是由於過量的1-磷酸果糖抑制了肝磷酸化酶所致。
此症是由於1-磷酸果糖醛縮酶(醛縮酶B)缺陷引起,病人肝內1-磷酸果糖醛縮酶活性幾乎完全缺失,而1,6-二磷酸果糖醛縮酶活性降低50%以上,造成肝內1-磷酸果糖的堆積及Pi和ATP的消耗。由於Pi大量消耗,肝粒線體氧化磷酸化減少,造成ATP缺乏。後者缺乏使肝細胞ATP依賴性離子泵功能障礙,膜內外離子梯度不能維持,細胞腫脹,細胞內容物外溢。

3.1,6-二磷酸果糖酶缺乏症此症為常染色體隱性遺傳病,多在嬰兒時發病。病兒表現為肌無力、嘔吐、嗜睡、生長停滯和肝腫大等,感染可促使急性發作。若不治療,在嬰兒期就可死亡。
實驗室檢查可見空腹血糖低,即空腹性低血糖、酮血症、乳酸血症和血漿丙氨酸水平增高。診斷依據低血糖症,確診需用肝、腎、腸活檢標本測定該酶活性。
治療主要通過食物療法,食含果糖少的食物,少吃多餐,避免飢餓,一般療效還可以,預後尚可。
5.半乳糖血症半乳糖在體內的正常代謝途徑如圖3-10所示。
影響半乳糖利用的各種因素均可引起半乳糖血症(galactosemia),但遺傳性半乳糖血症主要有兩種。
1-磷酸半乳糖尿苷轉移酶所致的半乳糖症是最多見的遺傳性半乳糖血症,其為常染色體隱性遺傳疾病。患者半乳糖代謝終止於1-磷酸半乳糖階段。雜合子病人此酶僅活性低下,但如果不持續服用半乳糖飲食,酶活性還可以維持健康。而純合子病人此酶完全缺乏,如食用富含半乳糖的食物,病人會出現嚴重病變,甚至死亡。如果及時停用含半乳糖的食物,病人除智力障礙外,其他的各種症状均可消失。此型半乳糖血症臨床表現為生長停滯,餵奶後嘔吐和腹瀉,繼而出現黃疸、溶血、肝大、智力障礙,檢查中可見患兒血清半乳糖水平明顯升高,尿中出現半乳糖,紅細胞中1-磷酸半乳糖尿苷醯轉移酶缺乏。患兒進食半乳糖或乳糖後,常伴有低血糖和高半乳糖血症。
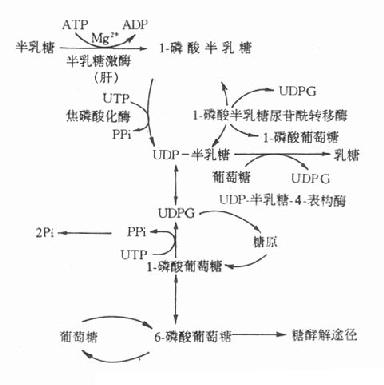
圖3-10 半乳糖代謝途徑
另一種遺傳性半乳糖血症是由於半乳糖激酶缺乏所致,此型症状較輕,新生兒期不表現症状,往往發生白內障後才被確診。
半乳糖血症的危害不是由於缺乏某種必需物質,而是由於產生毒性物質所致。半乳糖還原產物-半乳糖醇,在細胞內高濃度,如貯積於晶體,將吸收水進入晶體,造成晶體腫脹、混濁,引起白內障。此外1-磷酸半乳糖可能是某些毒性物質的前體,或者高濃度時本身就具有毒性作用。體外實驗已證實,1-磷酸半乳糖可抑制磷酸葡萄糖變位酶和G-6-P酶的活性。
(四)粘多糖沉積症
因蛋白聚糖降解酶先天性缺陷所引起的蛋白聚糖分解代謝障礙,將導致產生各種類型的粘多糖沉積症(mucopo-lysaccharidoses)。其特徵是過多的寡聚糖堆積與排泄。由表3-7可知,各型粘多糖沉積症的代謝基礎相似,但遺傳類型和臨床表現各不相同。
表3-7 粘多糖沉積症的酶缺陷
| 名稱 | 代號 | 酶缺陷 | 酶學測定樣品 | 生化改變 | 遺傳特性 |
| Hurler,Scheie症候群 | MPSⅠ | α-L-艾杜糖酸苷酶 | 成纖維細胞、白細胞、組織、羊水細胞 | 尿和組織中DS、HS增多,成纖維細胞中DS增加 | 常染色體隱 性 |
| Hunter症候群 | MPSⅡ | 艾杜糖醛酸硫酯酶 | 血清、成纖維細胞、白細胞、組織、羊水、羊水細胞 | 同上 | X連隱性 |
| Sanfilippo症候群A | MPSⅢA | HS-N-硫酸酯酶(硫醯胺酶) | 成纖維細胞、白細胞、組織、羊水細胞 | HS在尿中和組織中增多、DS在成纖維細胞中增多 | 常染色體隱性 |
| Sanfilippo症候群B | MPSⅢB | α-N乙醯葡萄胺苷 | 血清、成纖維細胞、白細胞、組織、羊水細胞 | HS出現於尿中 | 同上 |
| Sanfilippo症候群C | MPSⅢC | 乙醯基轉移酶 | 成纖維細胞 | HS出現於尿中 | 同上 |
| Morquio症候群 | MPSⅣ | N-乙醯半乳糖胺-6-硫酸酯酶 | 成纖維細胞 | KS和CS出現於尿中 | 同上 |
| Morquio症候群 | β-半乳糖甘酶 | 成纖維細胞 | KS出現於尿中 | ||
| Maroteaux-Lamy症候群 | MPSⅥ | N-乙醯半乳糖胺4-硫酸酯酶(芳香硫酸酯酶B) | 成纖維細胞、白細胞、組織、羊水細胞 | DS出現於尿中 | 同上 |
| β-葡糖醛酸苷酶缺乏症無名疾病 | MPSⅦ | β-葡糖醛酸苷酶 | 血清、成纖維細胞、白細胞、羊水細胞 | DS、HS(±)出現於尿中 | 同上 |
| 無名疾病 | MPSⅧ | N-乙醯葡糖胺6-硫酸酯酶 | 成纖維細胞 | KS和HS(±)出現於尿中 | 同上 |
註:MPS-粘多糖沉積症DS-磷酸皮膚素
KS-硫酸角質素
遺傳性粘多糖沉積症約佔出生嬰兒的1/30000。
| 關於「臨床生物化學/其它糖代謝異常」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |