臨床生物化學/糖分解代謝途徑的先天異常
| 醫學電子書 >> 《臨床生物化學》 >> 糖代謝紊亂 >> 糖代謝的先天性異常 >> 糖分解代謝途徑的先天異常 |
| 臨床生物化學 |
|
|
糖分解代謝途徑先天代謝異常可有丙酮酸激酶缺乏病,丙酮酸脫氫酶缺乏症和磷酸果糖代謝異常所致惡性發燒。
(一)丙酮酸激酶(PK)缺乏病
在糖酵解過程中,丙酮酸激酶催化磷酸烯醇式丙酮酸生成烯醇式丙酮酸,同時產生ATP,是酵解途徑產生ATP的反應之一,PK缺乏將導致成熟紅細胞缺乏ATP,進而發生溶血。
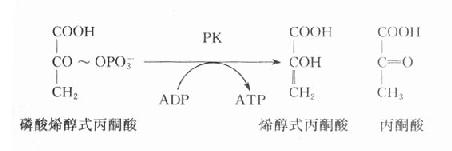
表3-6 各型糖原貯積病的特徵
| 類型 | Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | Ⅵ | Ⅶ | Ⅷ | Ⅸa | Ⅸb | Ⅹ | 0 | |
| 受害器官 | 肝腎 | 所有組積 | 肝、肌、心 | 肝單核吞噬細包系統 | 肌肉 | 肝 | 肌肉 | 腦 | 肝 | 肝 | 肝、肌肉 | 肝 | |
| 糖原結構 | N | N | 短枝異常 | 長枝異常 | N | N | N | 巨大顆粒 | N | N | |||
| 酶缺陷 | G-6-P酶 | α葡萄糖 苷酶 | 脫枝酶 | 分枝酶 | 肌磷酸化酶 | 肝磷酸化酶 | 磷酸果粒糠酶 | 肝磷酸化酶 | 同左 | 糖原合成酶 | |||
| 空腹低血糖 | ++ | 0 | + | 0 | 0 | ± | 0 | 0 | ± | 0 | ++ | ||
| 對胰高血 糖素反應 |
血糖 | 0 | ↑N | 0↑ | ↑ | ↑ | 0± | ↑ | ± | N進餐0空腹 | |||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 半乳糖 果糖試驗 |
血糖 | 0 | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ||||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 劇烈運動後血乳酸 | ↑ | ↑ | 0 | 0 | ↑ | ↑ | ↑ | ||||||
| 脂質代謝 | ↑↑ | N | 餓後 FFa ↑ |
運動後肌攝取 FA↑ |
TG↑ Ch↑ |
TG↑ Ch↑ |
TG↑ Ch↑ |
||||||
| 其它診斷性試驗 | 肝活檢 | 紅細胞抗原 ↑ | 紅細胞糖原結構異常 | 紅細胞磷酸化酶↓ | |||||||||
| 臨床特徵 | 生長停滯,肝大 | 心衰 | 似Ⅰ型但不明顯 | 肝硬化腹水 | 肌無力 | 肝大 | 肌無力 | 意識障礙 | 肝大,常染色體隱性遺傳 | 肝大伴性遺傳 | 肝大 | 輕度肝大 | |
註:N-正常;O-陰性;↑-升高;↓降低;±-可疑
網織紅細胞中含有粒線體,故可通過糖有氧氧化產生足量的ATP。而成熟紅細胞中不含粒線體,完全依賴糖酵解供能。紅細胞內生成的ATP主要用於維持細胞內外的離子梯度,特別是通過Na+-K+-ATP酶維持細胞內外Na+、K+濃度梯度。這對於維持紅細胞雙凹形狀十分重要。若缺乏ATP,紅細胞將發生腫脹,易發生溶血,實驗室檢查可以見到自身溶血試驗陽性。PK的遺傳缺陷是糖酵解途徑中遺傳性缺陷導致溶血性貧血的最多見原因。PK缺陷時,細胞中PK活性僅為正常細胞的5%-25%,故雖然PK缺陷少見,但其造成的溶血性貧血卻對機體危害甚大。
(二)丙酮酸脫氫酶複合物缺乏症
丙酮酸脫氫酶複合物由丙銅酸脫氫酶、二氫硫辛酸轉乙醯基酶、二氫硫辛酸脫氫酶及NAD+、FAD、CoASH、焦磷酸硫胺素、硫辛酸三個酶、五個輔助因子組成,其氧化的丙酮酸氧化脫羧生成乙醯輔酶A的反應是糖進入三羧酸循環、徹底氧化成CO2和水、產生大量ATP的關鍵。
在兒童中發現有多種丙酮酸代謝異常的疾病,其中有些是由於丙酮酸脫氫酶複合物中某些組份先天性缺陷所致。該酶複合物中各種亞基(催化亞基和調節亞基)都可能發生先天性缺陷。這些缺陷都可使丙酮酸不能繼續氧化產生ATP,使腦組織不能有效地利用葡萄糖供能,進而影響了兒童大腦的發育和功能,嚴重者可導致死亡。
丙酮酸不能進一步氧化,致使患兒血液中乳酸、丙酮酸和丙氨酸的濃度顯著升高,出現慢性乳酸酸中毒。丙酮酸脫氫酶的缺陷可以通過皮膚成纖維細胞培養並進行酶學測定予以測定。此類病人在一定程度上可通過進食生酮食物和限制糖的攝入使病情緩解或得到控制。
(三)磷酸果糖代謝異常
磷酸果糖激酶與果糖-1,6-二磷酸酶是作用相反的一對酶,它們所催化的化學反應是糖代謝途徑中的一處無效循環(又稱底物循環)。

由於酶的遺傳性缺陷,以上無效循環得不到控制,造成ATP大量分解產熱:ATP+H2O→ADP+Pi+熱。臨床上可因服用氟烷而誘發惡性發燒。
惡性發燒是一種罕見的遺傳缺陷性疾病,其發病率占兒童的1/15000,成人的1/50000-1/100000。病人常因服用某種藥物,如吸入氟烷而在幾分鐘內突然發病,表現為體溫驟然升高、代謝性和呼吸性酸中毒,以及高血鉀症和肌肉強直。人們認為,氟烷可以促進肌肉中上述兩個酶所催化的耗能無效循環,誘發惡性發燒的產生。
| 關於「臨床生物化學/糖分解代謝途徑的先天異常」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |