臨床生物化學/先天性代謝缺陷病
| 醫學電子書 >> 《臨床生物化學》 >> 遺傳性疾病的生物化學與分子生物學診斷 >> 常見遺傳性疾病的發生與基因診斷 >> 先天性代謝缺陷病 |
| 臨床生物化學 |
|
|
(一)苯丙酮尿症
苯丙酮尿症(PKU)因患者尿中含大量苯丙酮酸而得名病因是患者肝缺乏苯丙氨酸羥化酶,使由食物攝入體內的苯丙酮酸不能正常代謝為酪氨酸,導致血清中苯丙氨酸濃度升高,可高達50-100mg/dl(正常參考值為1-3mg/dl)。大量的苯丙氨酸使旁路代謝活躍,經苯丙氨酸轉氨酶作用生成苯丙酮酸。
苯丙氨酸羥化酶是在肝細胞中合成的,即在肝細胞中專一表達而在胎兒的絨毛細胞或羊水細胞中並不表達,給PKU的產前診斷帶來了困難。
由於診斷分子生物學技術的發展,1983年胡流清等人完成了人苯丙氨酸羥化酶cDAN探針的製備。他們利用探針和Southern blot技術進行分子雜交,通過限制性片段長度多態性(restriction fragment-length polymerphism ,RFLP)分析實現了PKU的產前基因診斷。他們先用MSP-I限制性內切酶消化正常人基因組DNA,經核酸電泳後,將DNA片段轉移到一張硝酸纖維膜(或尼龍膜)上,再用32p標記的人苯丙氨酸羥化酶cDNA探針雜交後,再經放射自顯影技術顯示正常人群有23kb和19kb兩種限制性片段長度。同時他們調查了一個PKU家系,進行比較發現,同樣用Msp-I內切酶消化,父母同時有23kb和19kb DNA,患兒只有19kb片段。說明在該家系中,苯丙氨酸羥化酶突變基因是與19kb片段連鎖的。連鎖簡而言之就是指同一條染色體上的基因聯合遺傳的現象。因此在該家系中的第2胎,如果只出現19kb DNA片段將是患兒,而同時出現23kb和19kb DNA片段或只出現23kbDNA片段的都是正常個體,而且只出現23kb DNA片段者不攜帶突變基因,不是突變基因的攜帶者。正常人出現19kb僅代表蛋白質和酶的多態。PKU家系的患兒為19kb,正常兒為23kb說明該PKU家系的基因突變與19kb連鎖。(表15-2)
表15-2 人苯丙氨酸羧化酶基因核酸電泳與cDNA探針雜交自顯影
| 正常人多態 | PKU家系 | |||||
| 父 | 母 | 患兒 | 正常胚胎 | |||
| 23kb | - | - | - | - | - | |
| 19kb | - | - | - | - | ||
1986年胡流清等進一步證明苯丙氨酸羧化酶的表達異常是基因點突變所致,並確定了突變點位置,並由此設計了正常和突變的各含21個核苷酸的寡核苷酸探針。
↓
正常探針5′TCCATTAACAGTAAGTAATTT3′
突變探針5′TCCATTAACAATAAGTAATTTT3′
↑
用這兩個探針分別與PKU家系成員的DNA雜交,證實與正常探針雜交者為正常個體,與突變探針雜交者為患者,同時可與兩個探針雜交者為正常個體,但攜帶突變基因。此法已成功用於產前診斷。(圖15-1)
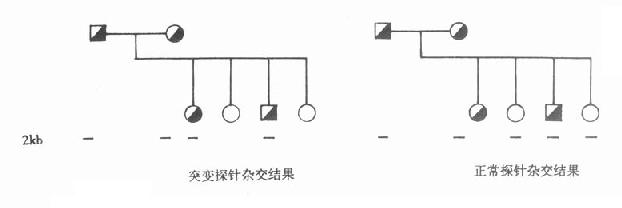
圖15-1 正常與突變苯丙氨酸羥化酶探針與PKU家系成員的DNA雜交結果
雜交技術由於費時、操作複雜而難以在基層實驗室推廣。現在多用聚酶鏈反應(PCR)擴增技術進行基因診斷,或在PCR基礎上再進行探針雜交,在提高靈敏度的同時又提高特異性,而且快速、經濟、簡便實用。
(二)嘌呤代謝紊亂與痛風
尿酸是嘌呤核苷酸分解代謝的重要產物,血和尿中尿酸濃度的檢測是嘌呤代謝紊亂的重要化學指標。可由經典的磷鎢酸還原法和高度特異的尿酸酶等化學方法檢測。
健康成年男子每天約生成尿酸600-700mg,其中60%-70%經腎排出,剩餘約200mg排入腸道由細菌降解。體內尿酸池維持在1200mg尿酸水平。腎排出尿酸的機制是由於尿酸分子量小(168u),可全部經腎小球濾過,但至少有98%被近曲小管重吸收,再經遠曲小管主動分泌,因此隨尿排出的尿酸主要由腎小管所分泌。腎每天約排出尿酸400-500mg(2.4-3.0mmol/L),相當於腎小球原濾液中含尿酸的4%-5%。
在血漿pH為7.4時,尿酸幾乎完全以單鈉尿酸鹽的形式存在,溶解度有限,約為0.42mmol/L(7.0mg/dl);而尿酸的溶解度更低,在pH5的尿液中,比尿酸難溶20倍。健康成年男子的血清尿酸濃度約0.3mmol/L,女子約低20%。血清尿酸水平隨年齡增加而增高,男性比女性更明顯,可達0.46mmol/L。血清尿酸超過0.42mmol/L為高尿酸血症(hyperuricemia)。在血清尿酸濃度超過尿酸鹽的溶解度時,就有可能尿酸鈉的針狀結晶沉澱於關節、肌腱、韌帶、腎錐體的間質組織等軟組織。足夠數量的尿酸鹽結晶可引起急性炎症反應,如沉積於關節腔,形成急性關節炎。這是由於尿酸鹽結晶被白細胞吞噬後,破壞溶酶體膜,使膜內酶釋放損傷白細胞和周圍組織,引起關節炎症状。表現為關節劇烈疼痛,反覆發作,此即痛風(gout)。沉積於軟組織的結晶稱為痛風石,周圍發生炎症反應則構成痛風結節。原發痛風可由於遺傳所致的核苷酸代謝中某一種酶表達異常所致,繼發性痛風則繼發於多種疾病。繼發性病因可能有大量服用葡萄糖、果糖與甘露糖,使體內嘌呤合成增加;或多發性骨髓瘤、紅細胞增多症、惡性貧血、牛皮癬和廣泛轉移的惡性腫瘤,使核蛋白轉換率增快,而尿酸生成過多;或細胞毒藥物或放射治療時,核酸分解亢進使尿酸鹽進一步增高。
先天性遺傳原發通風病因是次黃嘌呤-鳥嘌呤磷酸核糖轉移酶(HGPRT)缺失所致,部分缺失時,臨床表現為尿酸過多的痛風特徵;當完全缺失時,表現為高尿酸血症、精神發育遲緩等特徵的自毀容貌症候群。
HGPRT蛋白質分子為相同亞基的四聚體,每個亞基由217個胺基酸殘基組成。HG-PRT基因定位於X染色體長臂遠端,因此該病表現為X連鎖。HGPRT基因長約34kb,而成熟的mRNA的長度只有1.6kb,可見DNA分子內含有大量的內含子。HGPRT的突變基因及表達產物(mRNA,蛋白質)已被分離、研究,和血紅蛋白一樣有許多突變類型,如有的109位絲氨酸被亮氨酸代替,有的103位絲氨酸被精氨酸代替,還有報告基因發生缺失、重排的嚴重發病患者。1983年Wilson等發現一例DNA上的TagI限制性內切酶的切點發生突變,使在正常情況下可切出2.0kb片段的電泳區帶,由於切點突變而失去酶切作用,變成4.0kb的區帶出現。在蛋白質水平進一步研究發現50位的精氨酸被甘氨酸取代。
| 關於「臨床生物化學/先天性代謝缺陷病」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |