急診醫學/心臟驟停後的病理生理變化
| 醫學電子書 >> 《急診醫學》 >> 總論 >> 心、肺、腦復甦 >> 心臟驟停後的病理生理變化 |
| 急診醫學 |
|
|
一、體內各種主要臟器對無氧缺血的耐受力
正常體溫時,心肌和腎小管細胞的不可逆的無氧缺血損傷閾值約30min。肝細胞可支持無氧缺血狀態約1~2h。肺組織由於氧可以從肺泡彌散至肺循環血液中,所以肺能維持較長一些時間的代謝。
腦組織各部分的無氧缺血耐受力不同,大腦為4~6min,小腦0~15min,延髓20-30min,脊髓45min,交感神經節60min。
促使細胞發生不可逆的死亡機制,目前還只是些概念和假說,尚未形成一整套的理論。
二、無氧缺血時細胞損傷的進程
心臟驟停後,循環停止,如立即採取搶救措施,使組織灌流量能維持在正常血供的25%~30%。大多數組織細胞和器官,包括神經細胞均能通過低氧葡萄糖分解,獲得最低需要量的三磷酸腺甙(ATP)。心臟搏動的恢復性很大,腦功能也不會受到永久性損傷。如血供量只達15%~25%之間,組織細胞的葡萄糖供應受到限制,氧亦缺乏,ATP的合成受到嚴重影響,含量降低。如心臟搏動未恢復,組織灌流量亦未能增加,ATP就會耗竭,正常細胞的內在環境穩定性即被嚴重破壞。此時如再加大組織灌流,反而會促使組織細胞的損傷達到不可逆的程度,即所謂「再灌流所致的損傷」。
如組織灌流量在心臟驟停後,只維持在正常血供的10%以下,即所謂的「涓細血流」,ATP迅速耗竭,合成和分解代謝全部停頓,稱為「缺血性凍結」。此時蛋白質和細胞膜變性,粒線體和細胞核破裂,胞漿空泡化,最後溶酶體大量釋出,細胞發生壞死。這是一幅細胞不可逆變化的景象。
70年代末,Hearse和Nayler等提出缺血性心肌在某種條件,再灌流反而損壞了有可能恢復的心肌細胞。這被認為是再灌流損傷造成的細胞死亡,應該與缺血所致細胞死亡的概念區分開來。心臟驟停後,組織灌流立即停止,並不立即死亡。前面已提到,不同組織細胞的無氧缺血耐受閾值不同。那麼究竟是心肌細胞本身由於長時間缺氧缺血,已經發生了嚴重損傷,而再灌流帶來了多種有害物質,於是加速細胞死亡;抑或再灌流所帶來的有害物質,如大量的鈣離子、氧游離基、雙價鐵游離子等等。使本有可能恢復的缺氧缺血細胞完全失去恢復的能力。這似乎是一個矛盾現象:心臟驟停,組織灌流停,必須使之立即恢復,重新給細胞帶來所需的氧,恢複合成ATP,提供能量,使細胞恢復功能。組織細胞如在無氧缺血耐受時限內,能獲得正常血供的25%~30%,就有希望使復甦成功。或使用鈣離子通道阻滯劑、氧游離基清除劑、鐵離子螯合劑於再灌流的血液中,有的學者已在實驗動物中取得防止再灌流損傷的作用。這是當前復甦學的一項重點研究課題。
三、鈣離子在無氧缺血時細胞損傷中的作用
正常情況下,細胞外和細胞內的Ca2+梯級差為10000:1。它的兩個主要作用是:
(一)延緩房室交界區的傳導和延長該區細胞的不應期 這可使左、右束支和心室肌纖維恢復極化,使下傳的脈衝可以順利地進行心室肌細胞除極,不致因遇到尚處於不應期的束支而影響傳導;同時因為在交界區的延緩,就有足夠時間讓心室充盈得較滿意。
(二)形成電和機械耦聯 結合肌動蛋白和肌凝蛋白,心肌和血管平滑肌方能收縮。
鈣離子進入細胞後,促發細胞內儲存庫(肌漿網)釋出儲存的Ca2+(圖6-1)。兩者的總量足夠提供細胞蛋白質收縮所需。多餘的Ca2+由ATP泵出細胞外。如ATP合成受阻,不能泵出多餘Ca2+至細胞外;同時由於細胞膜因無氧性缺血的影響,Ca2+同慢通道離子變成快通道離子,大量進入細胞內。細胞內的Ca2+濃度可以從0.1μmol的基數增高到接近細胞外的濃度1.0mmol。
細胞內增多的Ca2+儲存在粒線體內。Ca2+激活磷酯A2(一種破壞細胞膜完整性的酶)。細胞膜被破壞後,釋出花生四烯酸,是一種游離脂肪酸。再灌流時提供的氧,在環氧化酶催化下,生成大量血栓素(圖6-2),是強力的可使心肌纖維和血管壁平滑肌纖維攣縮物質,此外血栓素並破壞粒線體的膜。ATP主要在粒線體內合成,粒線體被破壞後,ATP不能合成,體內的能量就更易耗竭,到了不可逆的階段。
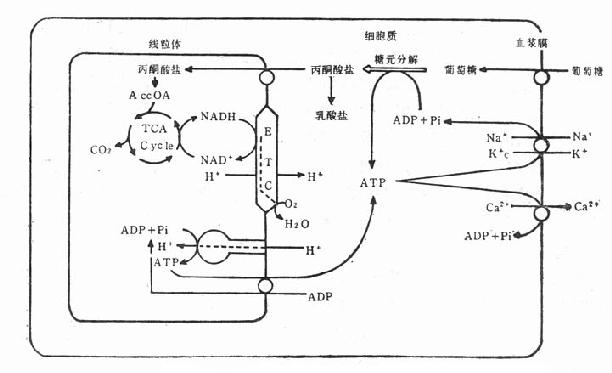
圖6-1鈣離子在心肌收縮的作用
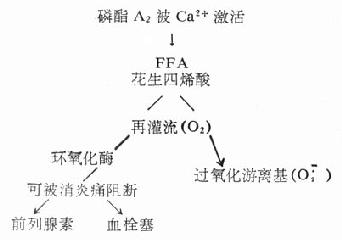
圖6-2 Ca2+激活磷酯A2示意圖
磷酯A2被激活後,釋放出遊離脂肪酸(FFA),細胞質中花生四烯酸含量增高。超過組織器官無氧缺血的耐受閾值之後,組織再灌流時,即產生一系列有害過程。
(引自Ann Emerg Med 12:471,1983)
四、氧游離基在組織無氧缺血時的破壞作用
氧是代謝作用必不可缺的因素。正常時,它在組織系統中經細胞內的色素系統作用,進行4價還原。在還原時,有1%~2%的氧分子逸出,進行單價還原,它具有高度反應作用的活性。因為單價還原的氧分子最外圈只含有一個離子,成為氧游離基,包括過氧化游離基(O ![]() )和氫氧游離基(
)和氫氧游離基(![]() )均屬極強的氧化或(和)還原物質。如果過多地存在,就會威脅細胞的完整性。正常時,由過氧歧化酶(superoxide dismutases,SODs)阻止這些游離基的過強作用。無氧缺血時,氧游離基含量在細胞內大量增加,超過氧歧化酶的清除作用,嚴重地破壞蛋白質和脂肪的成分,引起了廣泛的脂肪過氧化酶的連鎖反應,從而嚴重地破壞了細胞的正常結構。
)均屬極強的氧化或(和)還原物質。如果過多地存在,就會威脅細胞的完整性。正常時,由過氧歧化酶(superoxide dismutases,SODs)阻止這些游離基的過強作用。無氧缺血時,氧游離基含量在細胞內大量增加,超過氧歧化酶的清除作用,嚴重地破壞蛋白質和脂肪的成分,引起了廣泛的脂肪過氧化酶的連鎖反應,從而嚴重地破壞了細胞的正常結構。
五、鐵離子在組織無氧缺血時的破壞作用
上面提到缺血組織中,過氧化游離基含量過多,通過它的促發作用,引起鐵離子催化的Haber-Weiss反應,產生反應力極強的氫氧基。

粒線體中細胞色素,鐵蛋白(Ferritin)以及其他含鐵酶可以釋放足夠的游離的離子鐵進行催化作用。結果摧毀了細胞膜。而鐵螯合劑——去鐵胺(deferoxamine)可以起到保護作用。在實驗動物中,證實用去鐵胺(50mg/kg體重,靜脈注射)於心臟驟停(用注射冷1%氯化鉀使之驟停)的大鼠進行復甦時5min內使用,可以獲得100%的存活率。
六、腦復甦的重要性
近來對於心臟驟停後,神經系統受損的嚴重性和正確的治療方法已越來越引起臨床家的關注。一項臨床統計值得重視:經「復甦存活」而住院的病人,最終死亡,其中由於明顯的神經系統損傷者佔59%,因嚴重心力衰竭者佔31%。這些病人的組織損傷可以認為都是在再灌流以後加重的。有的學者稱之為「復甦後症候群」,大致可以分為三期:
(一)充血期 這是最初很短暫的時期,灌流可以超過正常時期,但是分布不均勻。目前尚不清楚這些增加了的血流是否確切灌注了微循環。
(二)低灌流期(或稱「無再灌流期」) 經過充血15~30min後,開始發生細胞水腫,同時出現血凝塊,紅細胞凝集,血流成泥流狀,血小板聚集。此外,還可能存在顱壓增高、腦血管收縮、毛細血管周圍紅細胞腫脹等。最終發生腦血管痙攣,此時腦血流顯著淤滯。這一低灌流現象在腦組織各部的嚴重程度並不一致,一般可持續18~24h。現在已經引起了臨床家和研究人員的嚴重關注,試圖改善這些異常現象,即在生命搶救方案中,增加適當防護措施,或在復甦術取得初步成功後,對這類病人加以特殊的強化監護治療。但是到目前為止,還沒有肯定的有效治療方法。
(三)後期 低灌流期以後,經過救治,腦組織可能部分恢復功能,並逐漸完全恢復(這與搶救時機及所採取的措施有密切關係);或持續性低灌流,導致長時間或永久性昏迷;或發展至腦死亡。
腦復甦後症候群全過程中的病理生理變化到目前為止和其他臟器在復甦過程中的病理生理變化一樣,所取得的都是零碎的、片斷的資料,還缺乏對它們的充分認識,亦尚未形成成套完整的理論體系。很多未知數尚待深入研究以求得明確的答案。有些情況是明了的。腦組織在人體器官中是最易受缺血傷害的。這是由於它的高代謝率、高氧耗量和對高血流量的要求。整個腦組織重量只佔體重的2%,但靜息時,它要求的氧占人體總攝取量的20%,要求的血流占心排出量的15%。心臟驟停後引起的無氧性缺血,腦組織中的ATP含量即減少90%。因此心搏停止後最早出現的症状之一是深昏迷。基礎生命搶救的主要目的亦即提供腦組織最低的血流量。
雖經實驗發現,心臟驟停後直至血供降至正常的30%~35%以下,大腦的神經元突觸的傳導功能都可以維持。如果降至20%以下,神經元的的生存力就受到損壞。但另有報導,如果大腦血供能維持在15%正常血供以上,並接受經過處理的再灌流血液:加入Ca2+通道阻滯劑、Fe2+螯合劑和清除氧游離劑的SODs、抗壞血酸、維生素E等,是有可能恢復腦組織功能的,但是至今還都是實驗室的結果,還沒有肯定的臨床實踐報導。
| 關於「急診醫學/心臟驟停後的病理生理變化」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |