醫學遺傳學/血紅蛋白病的分類和分子基礎
| 醫學電子書 >> 《醫學遺傳學基礎》 >> 單基因病 >> 基因突變致蛋白質合成異常 >> 血紅蛋白病 >> 血紅蛋白病的分類和分子基礎 |
| 醫學遺傳學基礎 |
|
|
|
1.異常血紅蛋白病 異常血紅蛋白(abnormalhemoglobin)是指由於珠蛋白基因突變導致珠蛋白肽鏈結構異常,如有臨床表現者稱為異常血紅蛋白病或異常血紅蛋白症候群。至今全世界已發現異常血紅蛋白471種。國內已發現60種,其中20種是世界首報。儘管異常血紅蛋白種類繁多,但僅約40%的異常血紅蛋白對人體有不同程度的功能障礙。
(1)異常血紅蛋白病的類型:
1)鐮形細胞病(sickle cell disease):此病主要見於黑人。該病系由於β鏈第6位谷氨酸被纈氨酸取代,形成HbS,導致電荷改變,在脫氧情況下HbS聚合形成長棒狀聚合物,使紅細胞鐮變,由於鐮變引起血粘度增高,導致血管梗阻性繼發症狀,一過性劇痛(肌肉骨骼痛、腹痛),急性大面積組織損傷,心肌梗塞可致死,鐮變細胞的變性降低還可引起溶血。HbS純合子(HbSHbS)表現為鐮形細胞性貧血,雜合子(HbAHbS)表現為鐮形細胞性狀,大部分無症状,但也可有輕度慢性貧血。
2)不穩定血紅蛋白病(unstable hemoglobinpathies):已發現的不穩定血紅蛋白在80種以上。由於Hb不穩定容易自發(或在氧化劑作用下)變性,形成變性珠蛋白小體(Heinz小體)。Heinz小體粘附紅細胞膜上,導致了離子通透性增加;另外,由於變形性降低,當紅細胞通過微循環時,紅細胞被阻留破壞,導致血管內、外溶血。不穩定Hb病一般呈常染色體顯性遺傳(不完全顯性),雜合子可有臨床症状,純合子可致死。臨床表現與Hb不穩定程度、產生高鐵血紅蛋白的多少以及不穩定Hb的氧親和力大小有關。輕者僅在服用磺胺等藥物或有感染時溶血;重者需反覆輸血才能維持生命。
3)血紅蛋白M病(HbM):HbM是因肽鏈中與血紅素鐵原子連接的組氨酸或鄰近的胺基酸發生了替代,導致部分鐵原子呈穩定的高鐵狀態,從而影響了正常的帶氧功能,使組織供氧不足,導致臨床上出現紫紺和繼發性紅細胞增多。本病呈常染色體顯性遺傳,雜合子HbM含量一般在30%以內,可引起紫紺症状。
4)氧親和力改變的血紅蛋白病:這類血紅蛋白病是指由於肽鏈上胺基酸替代而使血紅蛋白分子與氧的親和力增高或降低,致運輸氧功能改變。如引起Hb與氧親和力增高,輸送給組織的氧量減少,導致紅細胞增多症;如引起Hb與氧親和力降低,則使動脈血的氧飽和度下降,嚴重者可引起紫紺症状。
(2)異常血紅蛋白的分子基礎:異常血紅蛋白的發生涉及基因突變的各種類型,概括舉例如下。
1)單個鹼基置換:大多數異常血紅蛋白是由於珠蛋白基因發生單個鹼基置換所致,其中多為錯義突變。
①錯義突變:例如鐮形細胞貧血是β基因第6位密碼子GAG變成GTG。中國人較常見的HbE是β基因第26位密碼子由GAG(谷)→AAG(賴)所致。
②無義突變:例如HbMckees-Rock,其β鏈只有144個胺基酸組成,原因是β基因第145位酪氨酸密碼子TAT改變為終止密碼子TAA,使肽鏈合成提前終止。
③終止密碼突變:例如Hb Constant Spring就是由於α珠蛋白基因第142位終止密碼子TAA(mtRNA為UAA)突變為CAA(谷氨醯胺),結果α延長為172個胺基酸,這種突變基因轉形成的mRNAI不穩定,所以導致α鏈合成減少,表現為α+地中海貧血。
2)移碼突變:例如Hb Wagne是由於α鏈第138位絲氨酸密碼子UCC丟失一個C,致使其3』端鹼基順序依次位移,重新編碼,第142位終止信號變為可讀密碼,致使翻譯至147位才終止(圖4-13)。
3)整碼突變:例如Hb Gum Hiu是β鏈缺失第91-95胺基酸(亮-組-半胱-門冬-賴),但其前後胺基酸順序正常(圖4-13)。

圖4-13 人類α鏈和β鏈mRNAR不同突變類型
4)融合基因:例如Hb Lepore的類β鏈是由δ鏈和β鏈連接而成,肽鏈的N端像δ鏈,C端像β鏈,故稱為δβ鏈,相反,Hb反Lepore(Hbanti-Lepore)其N端像β鏈,C端卻像δ鏈,稱為βδ鏈。這是由於染色體的錯配聯會和不等交換而形成的融合基因(fusion gene)(圖4-14)。
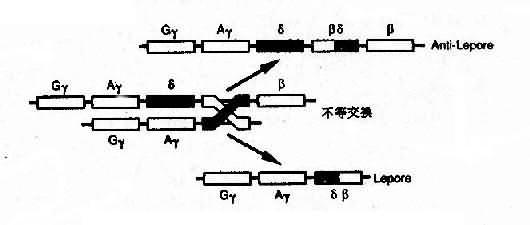
圖4-14 血紅蛋白融合基因形成機理
2.地中海貧血由於珠蛋白基因缺失或突變導致某種珠蛋的鏈合成障礙,造成α鏈和β鏈合成失去平衡面導致的溶血性貧血稱為地中海貧血(thalassemia)。根據合成障礙的肽鏈不同可把地中海貧血分為α和β地中海貧血兩類。此外還有少見的δβ和γβ地中海貧血。
(1)α地中海貧血(α-thalassemia,簡稱α地貧)是由於α珠蛋白基因的缺失或缺陷使α珠蛋白鏈(簡稱α鏈)的合成受到抑制而引起的溶血性貧血。如果一條16號染色體缺失1個α基因者稱α+地貧(亦稱α地2),缺失2個α基因者稱為α0地貧(亦稱α地1)。α地1的基因型可寫作―/αα,α地2的單倍型則寫成α-/αα.以上每種α地貧基因型與正常型配合可構成各種α地貧雜合子.各種α地貧基因型雜合子相互配合可構成各種純合子或雙重雜合子.α地中海貧血在我國多見於南方各省.
1)α地貧的臨床類型:根據臨床表現程度,依受累α基因數量不同而有差異,基本上可分為4類型.
①HbBart』s胎兒水腫症候群(HbBart』s hydrops fetalis syndrome):是兩條16染色體的4個α基因全部缺失或缺陷,基因型為α0地貧純合子(--/--),完全不能合成α鏈,不能形成胎兒HbF,相對過多的γ鏈形成γ四聚體(γ4)稱為HbBart』s(γ4).HbBart』s對氧親和力非常高,因而釋放給組織的氧減少,造成組織嚴重缺氧導致胎兒水腫,引起死胎或新生兒死亡.患兒血紅蛋白60%以上為HbBart』s,其餘為 HbPortland.
患兒父母均為α0地貧雜合子,基因型為α/--.他們若再生育,則胎兒有1/4的機會為αHbBart』s水腫胎兒,1/4為正常人,1/2為α0地貧雜合子(α地1)
②血紅蛋白H病:是α0地貧和α+地貧的雙重雜合子,即有3個α基因缺失或缺陷,基因型為-α/--或α-/--,也可為ααT/--(αT代表有突變,如Hb Constant Spring)。因缺失3個α基因,只能合成少量α鏈,β鏈相對過多,形成β四聚體(β4),易被氧化,導致β4解體成游離的單鏈,游離β鏈沉澱聚積包涵體,附著於紅細胞膜上,使紅細胞膜受損,失去柔韌性,易被脾破壞,導致中等度或較嚴重的溶血性貧血,稱為血紅蛋白H病(Hbh disease) .
患者雙親的基因型多為α0地貧雜合子(αα/--)和α+地貧雜合子(α/αα),或為α0地貧雜合子(αα/--)和非缺失型地貧雜合子(ααT/αα)。婚配後其子女有1/4機會為正常人1/4為α+地貧雜合子,1/4為α0地貧雜合子及1/4為HbH病。如父母一方有αT,則導致非缺失型HbH病。
③輕型(標準型 )α地中海貧血:為α0地貧雜分子(--/αα)或α+地貧雜合子(α/α-),缺失兩個α基因,間或有輕度貧血,我國主要是α0地貧雜合子。
輕型α地貧患者之間婚配,生育子女中可有1/4機會為HbBart水腫胎兒症候群。
④靜止型α地中海貧血:僅缺失一個α基因,為α+地貧雜合子(-α/αα),無症状。
靜止型α地貧與輕型地中海貧血個體婚配,可有1/4機會生育HbH病患兒。
2)α地中海貧血的分子基礎:從基因缺陷程度來區分,可把α地貧分為缺失型和非缺失型(點突變)。
①基因缺失:可分為α+和α0地貧兩種。α+地貧有兩種基因型:左側缺失(leftward deletion),缺失一個包括α2基因在內的DNA片段;②右側缺失(right ward deletion), 缺失範圍包括α2基因3』端和α1基因的5』端,結果形成了由α1的3』端和α2的5』端構成的融合基因。其發生機理是類α基因發生不等交換的結果。α0地貧基因缺失範圍差別很大(圖4-15)。

圖4-15 類α基因簇缺失類型
②非缺失型(點突變:(類型見表4-1)。
(2)β地中海貧血:β地中海貧血(β梩halassemia,簡稱β地貧),是由於β珠蛋白基因的缺失或缺陷使β珠蛋白鏈(簡稱β鏈)的合成受到抑制而引起的溶血性貧血。完全不能合成β鏈者稱β0地貧;能部分合成β鏈者(約為正常的5%-30%)稱β+地貧。此外,還有δβ地貧。它們可以有不同的組合,即β0地貧純合子(β0β0)、β0地貧雙重雜合子(β0/β+)、β0地貧雜合子(β0βA)、β+地貧純合子(β+/β+)和β+地貧雜合子(β+/βA)。β地貧在我國南方較常見。
1)臨床分類:大致可有4種主要類型。
①重型β地中海貧血:患者是β+地貧、β0地貧或δβ0地貧的純合子(其基因型分別為β+/β+、β0/β0和δβ0/δβ0)或是β+和β0地貧的雙重雜合子(基因型為β0/β+)。這些患者的β鏈幾乎不能合成,或合成量很少,以致無HbA或量很低,γ鏈的合成相對增加,使HbFt GbA2比率升高。由於HbF較HbA的氧親和力高,在組織中不易釋放出氧,所有β地貧患者有組織缺氧症狀。組織缺氧促使紅細胞生成素大量分泌,刺激骨髓的造血功能,使紅骨髓大量增生,骨質受鋟蝕致骨質疏鬆,可出現「地中海貧血面容」(頭顱大,額頂及枕部隆起,鼻樑塌陷,上頜及牙齒前突,眼距寬,眼瞼浮腫)。由於β鏈合成受抑制,過剩的游離α鏈形成α鏈包涵體,引起溶血性貧血,靠輸血維持生命。
表4-1 點突變引起的地中海貧血
| 分子缺陷類型 |
| 1、生成無功能或穩定性降低的 ①無義突變 a116GAG---TAG,(G---T) ②移碼突變 a130/31(--4bp) ③終止密碼突變 142TAA---CAA(T---C) 形成Hb Constant Spring ④起始密碼突變 a2ATG---ACG(T--C) 2、RNA加工突變 ①剪接改變 IVSI(GGTGAGGCT---GGCT) ②Poly(A)信號缺陷 AATAAAA ---AATAAG 3、產生不穩定H a2125CTG(亮)CCG(脯) 生成Hb Quong Sze |
②輕型β地中海貧血:患者是β+地貧、β0地貧或δβ0地貧的雜合子,基因型分別為β+/βA、β0/β+和δβ0/βA。這類患者由於還能合成相當量的β鏈,所以症状較輕,貧血不明顯或輕度貧血。本病特點是HbA2升高(可達4%-8%)或(和)HbF升高。
③中間型β地中海貧血:患者通常是某些β地貧變異型的純合子,如β+地貧(高F)/β+地貧(高F)或兩種不同變異型地貧的雙重雜合子,如β+,地貧/δβ+地貧。其症状介於重型和輕型之間,故稱為中間型β地中海貧血。
④遺傳胎兒血紅蛋白持續增多症:患者是由於β基因簇中某些DNA片段的缺失或者點突變,使δ和β鏈合成受抑制,而γ鏈的合成明顯增加,使成人紅細胞內HbF含量持續增多,故稱為遺傳性胎兒血紅蛋白持續增多症(hereditarypersistance of fetal hemoglobin,HPFH).HPFH的特點是HbF的成年人仍持續較高水平,無明顯的臨床症状。
2)β地中海貧血的分子基因:β地中海貧血迄今已發現100多種突變類型,其中10多種為缺失型,其餘均為點突變。我國已報導17種點突變。
點突變:絕大多數β地中海貧血是由於β基因發生點突變所致,突變涉及基因內及旁側表達順序的各個環節。主要類型有4種。
a.編碼區的無義突變、移碼突變和起始密碼突變:使生成的mRNA穩定性降低或形成無功能的mRNA,從而不能合成正常的β珠蛋白鏈,多數產生β0地貧,少數為β+地貧。例如無義突變密碼子17(A→G)、43(G→T)都產生β0地貧;稱碼突變41/42(-TCTT),71/71(+A)和β0地貧;以及起始密碼子突變ATG→AGG導致的β0都屬這類。見於中國人。
b.非編碼區IVS-1和IVS-2突變:影響前mRNA剪接等加工過程不能準確進行,形成異常的mRNA,導致β0或β+地貧。例如IVS-1的1位G→T為RNA拼接處改變;中國人中常見的IVS-2654,為內含子中由於鹼基置換形成了一個的裂解信號,影響正常位點的剪接,產生異常mRNA;還有一種是內含子中剪接位點的通用順序上的同義突變,從而激活內含子或外顯子中隱蔽裂解位點(cryptic splicihng site,CSS)如IVS-15(G→C)。CSS即DNA的一段順序在點突變後可以形成剪切識別順序(CCTATTGGT)的第7個鹼基G如變為A,則產生新的切點,即CCTATTAG↓T)。
c.影響轉錄的突變:這類突變主要集中於起始位點上游的啟動子TATA框,使轉錄效率降低,mRNA生成量減少而產生β+地貧。中國人的-29A→G,-28A→G都屬於這類。
d.RNA裂解部位缺陷:這類突變是由於異常的RNA加帽部位和多聚腺苷酸化信號的突變,從而影響RNA轉錄而不能準確裂解,產生不穩定的mRNA,使正常β鏈生成量減少,導致β+地貧。例如在mRNA加帽部位發生A→C顛換,引起β+地貧(亞洲人。)又如,多聚腺苷酸化信號AATAAA→AACAAA,引起β+地貧。
e.編碼區的外顯子突變引起剪接作用的改變:這類突變是由於編碼區的單鹼基突變(錯義突變或同義突變)激活鄰近的隱蔽裂解信號,影響IVS正常位點的剪接,產生異常的mRNA。如東南亞常見HbE,是一種輕型的β地貧,其原因是當β鏈26位密碼子發生G→A錯義突變時,其相鄰的裂解信號被激活,生成異常mRNA,產生HbE(圖4-16)。
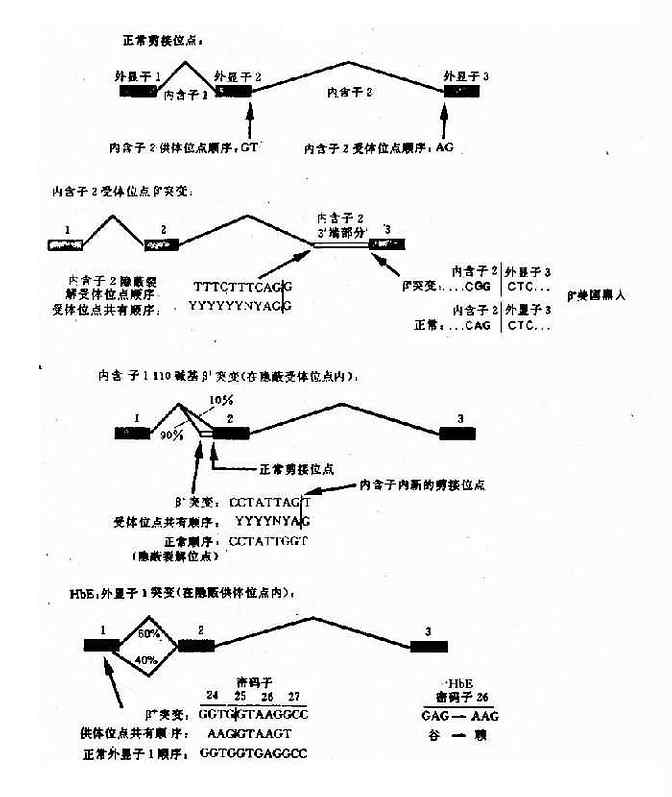
圖4-16 干擾正常β珠蛋白剪接的突變舉例
HbE:密碼子26(G-→A)
谷→賴
GAG→AAG(QAJ)(錯義突變)
β+(HbA 60%) βE(HbE 40%)
3)類β基因缺失:①按類β珠蛋白基因簇缺失長短大致可分為4種,即β0、δβ、γδ地中海貧血及遺傳性胎兒血紅蛋白持續增多症;②單純由於β0基因缺失引起的β地中海貧血罕見;③融合基因,如HbLeproe,是類β基因缺失7kb導致δβ融合基因,形成β0地貧。
| 關於「醫學遺傳學/血紅蛋白病的分類和分子基礎」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |