醫學遺傳學/受體蛋白病
| 醫學電子書 >> 《醫學遺傳學基礎》 >> 單基因病 >> 基因突變致蛋白質合成異常 >> 受體蛋白病 |
| 醫學遺傳學基礎 |
|
|
|
受體是存在於細胞膜上、胞質中或核內的一類具有特殊功能的蛋白質。現已證明具有調節生理功能和特異性受體達30種以上。其中包括多肽激素受體、固醇類激素受體以及神經遞質、前列腺素、免疫性因子、脂蛋白等受體。由於受體的本制是蛋白質,不言而喻,基因突變也可導致受體蛋白質和量的改變。由於受體蛋白遺傳性缺陷引起的一類疾病,稱為受體病(receptor diseae)。
受體病有獲得性與遺傳性之分。遺傳性受體病研究得較多的有下列4種:
1.家族性高膽固醇血症 家族性高膽固醇血症(familialhypercholesterolemia,FH)的臨床表現是:出生時即存在高膽固醇血症,增高的膽固醇主要為低密度脂蛋白膽固醇()LDL-C)和β極低密度脂蛋白(β-LDLC),黃色瘤(xanthoma)即增高的膽固醇在組織廣泛沉積。其最嚴重的後果是早年發生動脈粥樣硬化。純合子在5-30歲即表現心絞痛和心肌梗塞症状,可驟死。雜合子發生冠心病稍遲且發生率較低。
本病為常染色體顯性遺傳病。在人群中,雜合子發生率為1:500,雜合子臨床表現較輕,故屬不完全顯性遺傳,外顯率高(90%-100%)。
本病的主要病因是LDL受體(LDLR)的遺傳性缺乏。血漿中的LDL和β-VLDL與LDL受體結合後,內吞入細胞,在溶酶體中LDL與LDLR分離,LDLR回到細胞表面重新利用,LDL則在膽固醇酯酶作用下,釋出膽固醇供細胞利用。在這一過程中,細胞表面有功能的LDLR數量決定了血漿中LDL及β-VLDL的濃度。現知LDLR的編碼基因位於19p1.3-p13.3,總長度45kb,由18個外顯子組成。已發現FH患者有數十種突變,包括缺失型突變(主要的)、錯義突變、無義突變、移碼突變及整碼突變。表現為LDLR功能異常有4種類型:①細胞膜上完全無LDLR;②LDLR明顯減少;③LDLR無功能不能與LDL結合;④LDLR與LDL結合後不能內吞,可能是由於合成受體前後有加工缺陷。
2.睾丸女性化症候群 睾丸女性化症候群(testicularferminization syndrome)又稱雄性素全不敏感症候群(complete androgeninsensitivity syndrome,CAIS),為最常見男性假兩性畸形。患者核型為46,XY,有睾丸,能分泌雄性素,但缺乏雄性素受體,雄性素不能發生效應(圖4-20)。
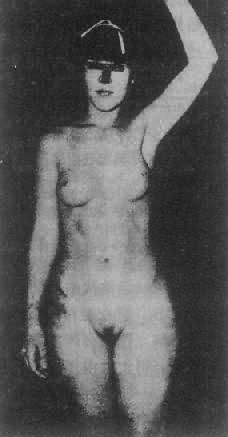
圖4-20睾丸女性化症候群患者(核型46,XY)
參看
| 關於「醫學遺傳學/受體蛋白病」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |