醫學遺傳學/凝血及抗凝血因子缺乏症
| 醫學電子書 >> 《醫學遺傳學基礎》 >> 單基因病 >> 基因突變致蛋白質合成異常 >> 凝血及抗凝血因子缺乏症 |
| 醫學遺傳學基礎 |
|
|
凝血是一複雜的生理過程,包括多種凝血因子及抗凝血因子參與。遺傳性凝血障礙包括低凝和高凝兩種情況。前者是由於基因突變致凝血因子活性降低,後者是由於抗凝血因子活性缺乏。
1.凝血因子缺乏症
除Ca2+與凝血活素外,所有凝血因子都有遺傳性缺乏的報告,其中血友病,特別是甲型血友病較為多見。現主要就血友病及與其有關的血管性假血友病加以介紹。
血友病(hemophilia)除甲、乙、丙三型外,加上後來又發現的一種vWF因子(vonwillibrand factor)缺乏的血管性假血友病,構成血友病的四種類型。1986-1989年全國24省、市,37個地區作血友病患病率的調查,調查人數16866 654人,總患病率2.73/10萬(男性5.21/10萬,女性0.06/10萬),與歐美相比較低,四型的構成比是:甲型79.8%,乙型14.1%,丙型2.8%,血管性假血友病3.3%。
(1)甲型血友病:甲型血友病(hemophilia A)又名抗血友病球蛋白(antihemophilicglobulin,AHG)缺乏症或第Ⅷ因子缺乏症。主要表現為出血傾向,其出血特點為:①緩慢持續滲血;②多發生於輕微創傷之後;③出血部位廣泛,常反覆發生,可形成血腫,並節變形,死因多為顱內出血。
現知,Ⅷ因子由三種成分組成:①FⅧ:C(AHG);②FⅧ:Ag(Ⅷ因子相關抗原);③vWF因子。甲型血友病為AHG遺傳性缺乏所致。
本病為X連鎖隱性遺傳,基因定位於Xq28,基因跨度超過186kb,由26個外顯子(佔9kb)及25個內含子(佔177kb)組成,編碼2351個胺基酸,已發現缺失型(包括錯義、無義及移碼突變等)共46種以上。雜合子的鑒定對開展遺傳諮詢很重要。過去多通過測定血漿AHG水平或用ⅧR:Ag/AHG比值來檢出雜合子,現已能採取分子遺傳學手段,特別是已成功地應用DNA印跡雜交、PCR技術等於產前診斷,這對防止重型患兒出生,十分有效。
治療可使用各種AHG製劑,但需長期使用,正在研究的基因治療將會是本病的根治方法。
(2)乙型血友病:乙型血友病(hemophilia B)又名血漿凝血活酶成分(PTC)缺乏症或第Ⅸ因子缺乏症。此型臨床表現酷似甲型,但發病率較低,遺傳方式亦為Ⅹ連鎖隱性遺傳,由於雜合子Ⅸ因子活性僅為正常1/3,某些雜合子可出現症状,故女性病人較甲型多見。
人類第Ⅸ因子基因已定位Xq27.1,基因總長度為34kb左右,由8個外顯子組成。已鑒定出的突變有100種之多(部分缺失及全缺失者30種,其餘為各種類型點突變)。我國王寧波等報告了重慶發現的5種點突變(外顯子2和5)。上海曾溢滔等發現1例-3內含子5kb片段缺失。在這些突變中發現幾例啟動子突變,臨床表現為兒童期有嚴重出血傾向,但到青春期後自發出血減輕。後證明,雄性類固醇可誘導啟動子產生Ⅸ因子,這也表明臨床症状輕重與突變性質可以有一定關係。
本型亦多採用輸血漿或濃縮血漿製劑治療,將第Ⅸ因子活性提高到25%以上即有療效。產前診斷是防止本病患兒出生的有效方法。近年,我國薜京倫等在Ⅸ因子缺乏症的基因治療方面取得了進展,有望在臨床取得長期穩定療效。
(3)丙型血友病:丙型血友病(hemophilia C)又名血漿凝血活酶前質(PTA)缺乏症(plasma thromboplastic antecedent deficiency)或第Ⅸ因子缺乏症。此型症状較甲、乙型輕。本病種族傾向明顯,多見於土耳其南部猶太人後裔。遺傳方式屬染色體隱性遺傳。現知,基因定位於15q11,基因長度為23kb,由15個外顯子組成,編碼625個胺基酸,但僅第11-15外顯子編碼羧基端具有凝血功能為Ⅺ因子主要成分。已發現3種點突變。本病純合子的Ⅺ因子活性在20%以下,雜合子為30%-65%。多數嚴重缺乏者用小劑量正常或濃縮血漿治療即顯效。
(4)血管性假血友病:血管性假血友病(von Willebrand disease)是一種較多見的與第Ⅷ因子有關的遺傳性凝血障礙。與本病有關的von Willebrand因子(v WF)是一大分子量的糖蛋白。基因定位於12pter-p12,vWF基因長度為180kb,有52個外顯子,mRNA總長度為9kb左右編碼2813個胺基酸。vWF蛋白由血管內皮細胞及巨噬細胞分泌,它在血中不僅作為Ⅷ因子載體,而且可增強Ⅷ穩定性,故vWF因子缺乏往往伴有Ⅷ因子活性降低。此外,血小板α顆粒中含vWF,故也參與血小板聚集,在凝血中發揮作用。本病患者有明顯的出血傾向。血中AGH活性降低,但不如甲型血友病嚴重,從基因水平已發現20多種突變型。
由於對本病基因有足夠的了解,已可通過RFLP連鎖分析或PCR法對本病進行產前診斷。
此外,由於遺傳性血小板缺乏或功能障礙和纖維蛋白原的遺傳性缺陷都可造成低凝狀態,在此不多贅述。
2.抗凝血因子缺乏症
(1)遺傳性抗凝血酶Ⅲ缺乏症:抗凝血酶Ⅲ(antithrombinⅢ,ATⅢ)對凝血酶Xa有抑制作用,肝素能加速其對凝血酶的抑制。其次,ATⅢ還有抑制Ⅸ、Ⅺ及Ⅻ的功能。
遺傳性抗凝血酶Ⅲ缺乏症(hereditary antithrombin Ⅲ deficiency)的臨床表現為容易發生血栓形成(主要部位為髂靜脈)及肺體塞。
本病為常染色體顯性遺傳。發病率在不同種族有顯著差異。歐美白種人中可高1:2000-5000。我國也已有個例報告。現知,ATⅢ基因定位1q23,基因長16kb,由7個外顯子組成,編碼432個胺基酸,至少已發現20種以上的突變類型,表現出不同的功能缺陷。
(2)遺傳性蛋白C系統異常:遺傳性蛋白C系統由蛋白C(PC)、蛋白S(PS)、血栓調節蛋白(thrombomdorlin,TM)及活性蛋白C抑制物(APC1)組成,它們之間的關係見圖4-19。活化的蛋白C有抑制Va、Ⅻa及促進纖維蛋白溶解的作用。蛋白C系統缺乏症有3種:
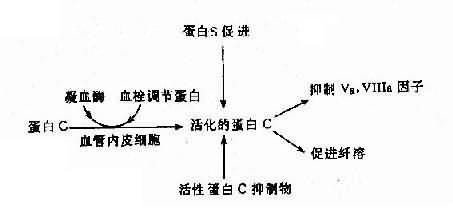
圖4-19 蛋白C、蛋白S及活性蛋白C抑制物的相互關係
1)蛋白C缺乏症:主要症状是靜脈血栓形成,常見於深部靜脈。本病發生率估計為1:16000,呈常染色體顯性(或不完全顯性)遺傳,純合子嚴重,表現為出血性皮膚壞死、瀰漫性血管內凝血(DIC)和血栓。雜合子多數在青壯年發病。蛋白C基因定位2號染色體,基因長12kb,由8個外顯子組成,已鑒定出若干種缺失型及點突變病例,也有涉及mRAN加工缺陷者。
2)蛋白S缺乏症:蛋白S的作用是促進活化蛋白C(APC)結合於磷脂,加速APC滅活Va因子。本病亦屬常染色體顯性(或不完全顯性)遺傳。基因定位在3號染色體,其總長度為45kb。上海也已報告2例PS缺乏症。
3)先天性活化蛋白C抑制缺乏症(congenitaldeficiency of activated protein Cinhibitor)。
其它還有遺傳性纖溶系統異常,包括①先天性異常纖溶酶原血症;②先天性纖溶酶原激活物釋放異常;③遺傳性纖溶酶原激活抑制物增多症;④血塊異常所致先天性纖溶減弱等,均表現出高凝狀態的各種症状。
| 關於「醫學遺傳學/凝血及抗凝血因子缺乏症」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |