病理生理學/原發性高血壓
| 醫學電子書 >> 《病理生理學》 >> 心力衰竭及高血壓 >> 高血壓 >> 高血壓的原因和機制 >> 原發性高血壓 |
| 病理生理學 |
|
|
原發性高血壓發生的原因和機制,尚不完全清楚,目前認為是多種因素參與的。
1.精神、神經因素 臨床觀察和動物實驗各方面的資料證明,本病的發生與精神應激和高級神經活動過度緊張有關:①長期從事與精神緊張有關的職業或處於過度精神應激的環境中的人,本病發病率高;②高血壓的發生與憂鬱、恐懼、悲傷等不良情緒有關;③給犬造成高級神經活動過度緊張狀態,可促進高血壓的發生和發展;④在本病早期,單純消除精神應激,常可使血壓恢復正常;使自發性高血壓大鼠(spontaneous hypertension rat, SHR)脫離應激環境,也可明顯推遲高血壓的發生。
精神應激引起高血壓的機缺可能是由於大腦皮層在各種精神應激長期作用下,通過興奮下丘腦神經內分泌中樞而使交感神經系統興奮,兒茶酚胺釋放增多。
已有許多事實證明,原發性高血壓時交感系統的活動加強;如①SHR大鼠內臟大神經放電率較正常鼠高;②某些本病患者,尤其是青年患者血漿中兒茶酚胺(主要是去甲腎上腺素)含量較高;③臨界高血壓時中樞和外周交感神經活動性都較血壓正常者高;④應用交感神經或α-腎上腺素受體阻滯藥可使血壓下降。
交感神經引起血壓升高的機制是多方面的:①使小動脈收縮,增大外周阻力;使靜脈收縮,增加回心血量;②通過興奮心臟的β受體使心臟收縮加強、加快,從而提高心輸出量;③直接或間接激活腎素-血管緊張素系統(renin-angiotensinsystem, RAS),進而收縮血管和通過血管緊張素Ⅱ(angiotensinⅡ,A-Ⅱ)促進醛固酮分泌,增加血容量(圖12-5)。
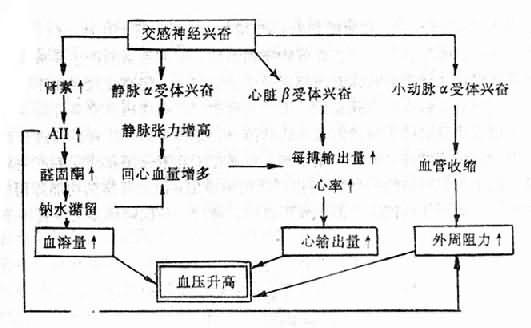
圖12-5 交感神經引起血壓升高的機制
但須指出,並非原發性高血壓患者血中兒茶酚胺的含量都升高;而且,即使升高,其程度(一般升高20~25%)也遠不足以導致高血壓。說明交感-兒茶酚胺系統的活動增強在原發性高血壓的發生中並非起決定性作用。現證明交感神經節後纖維有兩類,一類是以神經肽Y(neuropeptide Y, NAY)和去甲腎上腺素為遞質的縮血管纖維,另一類是以降鈣素基因相關肽(calcitonin gene related peptide, CGRP)和P物質為遞質的擴血管纖維。認為這兩類纖維的功能失衡即前者功能強於後者才是交感社經參與高血壓發生的重要機制。
精神-神經因素雖與高血壓發生有關,但並非唯一重要因素,因為遠非所有長期處於精神應激環境中的人都發生高血壓病;反之,發生高血壓病者也不一定有精神應激史。另外,由於本類因素引起的血壓升高多為一時性的,目前多數人認為精神-神經因素在原發性高血壓發生的始動機制中所起的作用較在維持機制中所起的作用為大。
2.腎素-血管緊張素系統(RAS)原發性高血壓時血漿腎素活性升高者佔20%,正常者60%,降低者10~20%。過去認為RAS只有在腎素升高組高血壓的發生中可能起一定作用,而在其他兩組不起作用。但晚近證明並不能排除RAS在其他兩組中的作用,因為:①應用RAS抑製藥,尤其是血管緊張素轉換酶抑製藥(angiotensin converting enzyme inhibitor ACEI)治療高血壓病時,不但對高腎素型有效,且對其它型都有效。目前已證明,除腎臟產生腎素影響血漿RAS水平外,心肌、血管平滑肌等組織也存在完整的局部RAS。血漿平滑肌的RAS也具較強的收縮血管加壓作用。高血墳時局部RAS活性升高並不影響血漿腎素的水平,但對高血壓的發生卻起著一定作用;②因高血壓病患者血管對加壓物質(包括血管緊張素)反應性增高,故即使腎素活性正常甚至低於正常時,對本病患者仍可能起著加壓作用。目前認為本系統在高血壓病尤其對高血壓的維持起著較重要的作用。
③鈉、鉀、鈣與高血壓臨床與實驗材料證明,食鹽與高血壓的發生有密切關係:例如①食鹽攝入量與本病發病率呈正相關,攝取食鹽多的地區(如日本本士和我國北京地區)發病率較食鹽較少地區(如阿拉斯加的愛斯基摩人和我國廣州地區)高;而牙買加某島上居民食鹽攝入量每人<2g/d,就無高血壓發生。②給SHR大鼠喂低鹽飲食,僅發生輕度高血壓;但給予高鹽飲食則迅速出現重度高血壓。③限制食鹽的攝入量或服用利尿藥增加排鈉量,對某原發性高血壓患者有較好的降壓作用。
關於食鹽引起高血壓的機制,尚不完全清楚,可能是多方面的:如①鈉瀦留導致細胞外液增加,從而可加大心輸出量,而且血管壁的鈉水溜留可使管腔狹窄,從而使外周阻力增大。②高鹽負荷可促使下丘腦產生利鈉因子(natriuretic factor, NF),又稱下丘腦抑制因子(hypothalamic inhibitor factor, HI)具有所謂利鈉激素樣的特性,為一種低分子量(500)的非肽類物質。它可抑制細胞膜Na+-K+-ATP酶的活性,故又稱Na+-K+-ATP酶抑制因子或鈉泵抑制因子。NF也可被看成是一種內源性洋地黃樣物質。這樣,NF一方面可降低腎小管上皮細胞對鈉的重吸收,引起利鈉效應;另方面使血管平滑肌細胞等細胞內的鈉瀦留起來,進而加強Na+-Ca2+交換,致使細胞內Ca2+增加,血管收縮(圖12-6)。③增高中樞和外周交感神經的活性;④高食鹽可使擴血管物質如激肽、前列腺素的產生釋放減少,使縮血管物質如血管緊張素產生增多並加強其與相應受體的親和力。但高食鹽引起高血壓是有條件的。高食鹽能否引起高血壓,關鍵在於腎臟能否將攝入過多的鈉排出。絕大多數人的腎臟通過排鈉利尿機制(食鹽過多,血Na+濃度增高,腎血容量增加,可通過①腎素分泌和醛固酮產生減少,和②腎內血壓增高等機制加強對鈉的排泄),可把過多的鈉排出;但當腎排鈉功能障礙時,由於不能把過多的鈉排出,即可導致鈉瀦留而出現高血壓。腎排鈉功能障礙多與遺傳因素有關。
流行病學調查證明,鉀的攝入量與血壓呈負相關;給高血壓患者補充鉀鹽可使血壓下降,並能提高限鈉飲食療法的降壓效果。鉀的降壓機制可能是:①抑制血漿腎素活性,減少血管緊張素Ⅱ受體數目和降低其與血管素Ⅱ的親和力;②抑制腎小管對鈉的重吸收,促進鈉排出;③減弱交感神經的敏感性。此外,高能尚能激活血管平滑肌細胞膜Na+-K+泵,防止鈉、鈣在細胞內蓄積([Na]i、[Ca]i)。
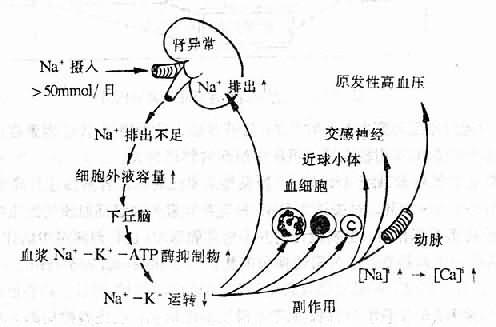
圖12-6 高食鹽和利鈉因子引起高血壓機制示意圖
據調查,某些原發性高血壓患者飲食鈣攝入量較正常人低,其血清Ca+和腎素水平也低,對這些患者長期補充鈣可使血壓降低,並能對抗高鹽所致的升壓效應。實驗也證明,減少飲食中的鈣,可使自發高血壓大鼠(SHR)血壓明顯升高;相反,如增加鈣攝入量,可延緩SHR幼鼠血壓的升高,並使SHR成年鼠血壓降低。鈣的降壓機制可能是:①穩定細胞膜結構,控制膜離子通透性,使Ca+不致大量進入細胞內;②鈣與鈣調蛋白結合,激活細胞膜鈣泵,增加鈣的外流,使細胞內鈣濃度降低。防止血管平滑肌細胞內Ca2+的積聚就可防止血管的收縮,從而防止血壓的升高。
4.遺傳因素已有充分證據說明高血壓病與遺傳基因有關:①有家族性:雙親都有高血壓病史的子女發病率為50%,即兩個子女一個發病;雙親之一有高血壓史的子女,發病率為25%;雙親沒有高血壓病史的子女,發病率為5%。②自幼收領的養子和親生子雖然生活環境完全相同,但後者的高血壓病發病率與雙親的發病率更為接近。③對食鹽敏感和不敏感的兩種大鼠,雖然在同樣高食的飼養條件下,前者發生高血壓而後者則否。
遺傳基因主要決定高血壓病發生的易感性,而非高血壓的本身。由於血壓受多種因素控制,故遺傳的「易感性」也是多基因決定的。其表現是多方面的。例如腎臟排鈉的先天性缺陷,細胞膜的先天性功能異常和血管平滑肌對加壓物質的敏感性高等,今分述如下:
(1)腎先天性排鈉缺陷:在同樣大量攝取食鹽的條件下,對鹽敏感鼠發生高血壓,而對鹽不敏感鼠則不發生高血壓。如把對鹽敏感鼠的兩個腎臟移植給對鹽不敏感鼠,又把對鹽不敏感鼠的兩個腎移植給對鹽敏感鼠,則對鹽不敏感鼠發生高血壓而對鹽敏感鼠不發生高血壓。這說明對敏感鼠所以發生高血壓,是由於腎排鈉障礙,而腎排鈉障礙是由遺傳因素決定的。人類高血壓病易感者也可能存在類似的遺傳性腎排鈉缺陷。
(2)細胞膜先天性功能異常:主要表現在膜的鈉、鈣離子運轉異常。細胞對鈉的運轉有四個系統,即①Ca+-K+泵:為主動的運轉系統;②被動性運轉,決定於細胞內外K+、Na+離子梯度;③Na+-K+同向運轉(co-transport)和④Na+-Li+逆向運轉(counter-transpot)(圖12-7)。
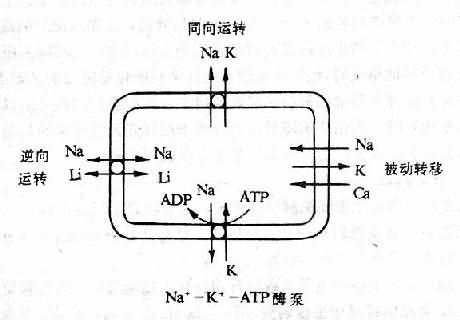
圖12-7 細胞內鈉運轉的四個系統
上述的③和④兩個運轉系統,雖不需ATP水解供能,但需在特異的載體參與下使相應的離子進行跨膜的易化擴散。這兩個系統都是協同運轉系統,即一種離子與載體結合後又可提高載體與另一種高子的親和力,進而使兩種離子偶聯協同擴散。除了Na+-K+泵的主動運轉外,Na+-K+同向運轉也是細胞用以防止細胞內Na+過度負荷的重要機制。Na+-Li+逆向運轉與腎臟近曲小管對鈉、水的重吸收呈正相關,即當Na+-Li+逆向運轉加強時,近曲小管對鈉、水的重吸收也加強。據報導,高血壓病患者和SHR大鼠上述四種鈉運轉系統都有可能發生改變,主要是Na+-K+泵運轉被抑制;同時,因膜通透性增高,故Na+被動轉入細胞增加;此外還有Na+-Li+逆向運轉加強和Na+-K+同向運轉減弱。其結果是細胞內鈉蓄積。研究結果也證明SHR大鼠和部分原發笥高血壓患者細胞內的鈉含量高於正常。細胞內鈉的蓄積。可以導致:①血管壁鈉水負荷增加,管壁增厚,血管腔狹窄,從而引起外周阻力增高;②Na+-Ca2+交換加強,使血管平滑肌細胞內Ca2+增加(可能還有其它機制),從而促使血管收縮。尤其是在管壁增厚的情況下,輕度的血管收縮就可產生較大的阻力。此外,由於機體的鈉水瀦留,又可促使利鈉因子的分泌和釋放,通過抑制Na+-K+泵,進一步加重鈉在細胞內蓄積。
在雙親有高壓病史而血壓正常的子女中,約有一半有細胞膜對鈉運轉的異常。
原發性高血壓患者細胞內Ca2+增多,除繼發於細胞內鈉升高外,還與膜對鈣的運轉系統異常有關,主要表現在:①SHR大鼠和原發性高血壓者紅細胞和血管平滑肌細胞膜對Ca2+的通透性增加,故Ca2+內流加強:②質膜對Ca2+的結合力決定於細胞膜表面多種Ca2+結合蛋白的量及其與Ca+的親和力。SHR大鼠和原發性高血壓患者可能由於結合蛋白量減少和/或親和力降低,從而導致膜結合鈣減少而胞漿中的游離鈣增加;③ATP依賴性鈣泵運轉障礙:現證明SHR大鼠的血管平滑肌肌膜、心肌肌漿網以及紅細胞等都有ATP依賴性鈣泵的運轉障礙,從而不能把胞漿中的Ca2+分別轉運到細胞外和肌漿網中,結果使Ca2+在胞漿中增加。血管平滑肌細胞內游離鈣的增多,被認為是原發性高血壓發生機制的最後共同途徑。現認為膜對鈉、鈣離子運轉的障礙是遺傳因素決定的膜功能異常的表現,而高血壓病可能是一種細胞膜病。
此外,實驗還證明SHR大鼠出生後血壓升高之前,其血管壁的收縮成分較正常鼠多,對加壓物質的收縮反應也遠較正常鼠強。雙親有高血壓病史的血壓正常子女也存在有類似的現象。同時這類子女血中的利鈉因子(NF)也明顯升高,這提示血管的高反應性和NF的升高都與遺傳有關。總之,高血壓病遺傳的易感性是多因素的,它在高血壓病發生中的作用是和環境因素的作用相輔相成的,也即環境因素往往是在遺傳易感性的基礎上發揮致病作用的,而遺傳易感性又是通過對環境因素的反應表現出來。例如食鹽過多之所以能引起鈉水瀦留和血壓升高,常是在腎臟先天性排鈉障礙和膜對離子運轉先天性異常的基礎上發生的,而患者對高血壓病的遺傳易感性又是通過食鹽過量才得以表現出來。
參看
| 關於「病理生理學/原發性高血壓」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |