臨床生物化學/光譜分析的常用方法
| 醫學電子書 >> 《臨床生物化學》 >> 常用分析技術在臨床生物化學中的應用 >> 光譜分析技術的應用 >> 光譜分析的常用方法 |
| 臨床生物化學 |
|
|
|
(一)吸收光譜分析法
1、可見光及紫外光分光亮度法
(1)標準曲線法:將一系列濃度不同的標準溶液按照一定操作過程顯色後,分別測吸亮度,以吸亮度為縱坐標,濃度為橫坐標繪製標準曲線。在相同條件下處理待測物質並測定其吸亮度,即可從標準曲線找出相對應的濃度。
(2)對比法:將標準樣品與待測樣品在相同條件下顯色並測定各自的吸亮度。由於測定體系溫度、厚度以及入射光波長是一致的,所以標準與待測樣品K值及L相等,可應用下式比較計算待測樣品濃度:Cx=Cs×As/Ax。
(3)差示法:有色溶液濃度太濃或太稀(透光度超過90%-10%)時,測定結果會產生較大誤差,此時可採用差示法(differentialspectrophotometry)。①高濃度樣液差示法:用標準品製備濃度稍低於試樣的參比溶液,先將儀器光門關閉,調節T%=0,再將參比溶液置於光路上,打開光門使T%=100,然後測定樣品溶液的透光率即可。例如某一樣品溶液原來的透光率讀數為5%(10%以下),用差示法後讀數為50%,這實質是把透光率標尺擴展了10倍,從而減少了測量誤差。②低濃度樣液差示法:用標準品製備濃度稍高於試樣的參比溶液,將它放在光路上,打開光門,調節透光率至0%,然後換以空白溶劑,調節刻度至100%。此後測定樣品的讀數即可。
(4)多組分混合物的測定:當試樣中有兩種或兩種以上的組分共存,可根據各組分的吸收光譜的重疊程度,選用不同的定量方法。如果混合物各組分的吸收峰互不干擾,這時可按單組分的測定方法,選擇測定波長,分別測定各組分的含量;若各組分的吸收峰相互重疊,可採用解聯立方程法、等吸收點法、雙波長法等解決量中的干擾問題。
(5)利用摩爾吸光係數進行檢測:①吸光係數:Beer定律的數學表達式為A=kbc,若溶液的濃度c以g/L為單位,b為光徑以cm為單位,則常數K稱為吸光係數,以a表示,其單位為升/(克.厘米)[L/(g.cm],A=kbc可寫成A=abc。②摩爾吸光係數:公式A=kbc中的以為1mol/L,b為1cm時,則係數k稱為摩爾吸光係數,以ε表示,單位為升/(摩爾.厘米)[L/(mol.cm)],A=kbc可寫成A=εc。在實際工作中,不能直接用1mol/L這種高濃度的溶液測定吸光度,而是在稀釋成適當濃度時測定吸光度進行運算。ε值與入射光波長、溶液的性質等因素有關。如NADH在260nm時ε為15000,寫成ε260NADH=15×103;在340nm時ε為6220,寫成ε340NADH=6.22×103。③比吸光係數:如公式A=kbc中的c是百分濃度(w/v)b為cm,則常數k可用E%表示,稱為比吸光係數或百分吸光係數,A=kbc可寫成A=E%bc。當待測物的化學結構是已知者可用ε值分析,若所測物的化學結構是未知的,則ε無法確定,此時用比吸光係數分析就很方便。a、ε和E常用作粗定量分析,主要用於定性分析。
近年來由於新的靈敏度高、選擇性好的顯色劑和掩蔽劑不斷出現,一般不經分離過程即可直接比色測定。幾乎所有的無機離子和有機物都可直接或間接用可見光及紫外光分光光度法進行定量測定。
2、原子吸收分光光度法 原子吸收光光度法是基於元素所產生的原子蒸氣中,待測元素的基態原子,對所發射的特徵譜線的吸收作用進行定量分析的一種技術。具有靈敏度高、選擇性好、操作簡便、分析速度快等優點,對大部分元素,其靈敏度約為10-8~10-10g/ml,是微量元素檢測的一個十分有鏟的方法之一。
(1)分析條件的選擇
1)分析線:原子吸收法中常選擇待測元素的共振作分析線,但並不是任何情況下都應使用共振線,例如As、Se、Hg的共振線在遠紫外區,該區域火焰吸收強烈,不宜選用共振線作分析線。在選擇分析線時,首先掃描空心陰極燈的發射光譜,然後噴入試樣溶液,觀察譜線的吸收和干擾情況,一般選用不受干擾且吸收最強的譜線作為分析線。常用元素分析線見表16-1。
表16-1 常用元素分析線
| 元素 | 分析線 | 元素 | 分析線 | 元素 | 分析線 |
| Al | 3093.3082 | Ca | 4227.2309 | Cd | 2288.3261 |
| Cu | 3248.3274 | Fe | 2483.3523 | Hg | 2537 |
| K | 7665.7699 | Mg | 2852.2796 | Na | 5890.3303 |
| Pb | 2167.2833 | Sn | 2246.2796 | Zm | 21393.3076 |
2)狹縫寬度的選擇:適宜狹縫寬度可由實驗確定:將試樣噴入火焰,調節狹縫寬度,測定不同狹縫寬度時的吸光度,達到一定寬度後,吸光度趨於穩定,進一步增加狹縫寬度,當其他譜線或非吸收透過狹縫時,吸光度立即減小。不引起吸光度減小的最大狹縫寬度,就是最適宜的狹縫寬度。
3)原子化條件的選擇:對於火焰原子化法,火焰的種類和燃助比的選擇是很重要的。當燃氣和助燃氣選擇好後,可通過下述方法選擇燃助比:固定助燃氣流量,改變燃氣流量,測量標準溶液在不同燃助比時的吸光度,繪製吸光度-燃助比關係曲線,以確定最佳燃助比。
對於石墨爐原子化器的使用。應注意乾燥是一個低溫去溶劑的過程,可在稍低於溶劑沸點的溫度下進行。灰化是為了破壞和去除試樣基體,故在保證試樣無明顯損失的前提下,將試樣加熱到儘可能高的溫度。原子化階段應選擇最大吸收信號的最低溫度。總之,根據試樣的性質確定各階段所選定的溫度與加熱時間。
4)試樣量的選擇:火焰原子化法在一定範圍內,噴入的試樣量增加,原子吸光度增大,但在超過一定量後,由於試樣不能完全有效地原子化及試液的冷卻效應會使吸光度不再增大甚至有所下降。因此在保持一定的火焰條件下,測定吸光度隨噴入試樣量的增加達到最大吸光度時的噴霧量,就是適宜的試樣量。石墨爐原子化一般固體取樣0.1~10mg,液體取樣量為1~50μl,主要依石墨管容器的大小而定。
(2)定量方法:常用的定量方法有標準曲線法、標準加入法和內標法。
1)標準曲線法:同紫外可見標準曲線法。但由於燃氣流量和噴霧效率的變化,單色器波長的漂移等因素可導致樣品測試條件與標準曲線測定條件不同,所以,在測定未知樣品時,應隨時對標準曲線進行檢查,每次實驗都要重新製作標準曲線。
2)標準加入法:在標準曲線法中,一般情況下要求標準溶液和未知溶液的組成保持一致。但在實際工作中不是總能做到的,採用標準加入法可以克服這個缺點。本法把未知試樣溶液分成體積相同的若干份,留其中一份,其餘分別加入不同量的標準樣品,然後測定各溶液的吸光度,以吸光度為縱坐標,標準樣品加入量為橫坐標繪製標準曲線(圖16-1)。由於未知樣品中含待測組分,故直線不通過原點而是在吸光度高於原點的A0處與縱軸相交,且直線外推法使工作曲線延長交橫軸處於B點,則OB所對應的濃度Cx就是未知試樣中待測組分濃度。
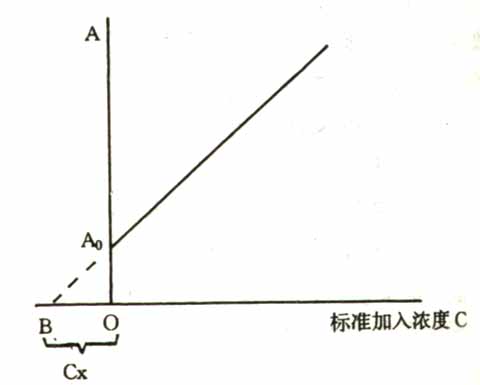
圖16-1 標準加入法工作曲線
標準加入法的優點是能夠更好地消除樣品中其它成分對測定的影響。
3)內標法:在系列標準樣品和未知樣品中加入一定量試樣中不存在的元素(內標元素),然後測得As和Ax,以As/Ax比值對標準樣品中待測元素濃度C繪製標準曲線,再根據未加內標元素的試樣A與加入內標元素的試樣As比值(A/As)即可在標準曲線上求得試樣中待測元素濃度。
本法要求內標元素應與待測元素有相近的物理和化學性質。此外,內標法只適用於雙通道型原子吸收分光光度計。
(二)發射光譜分析法
1、熒光分析法 許多物質都是光致發光的,即它們可以吸收電磁輻射,然後又重新發射出相同或較長波長的輻射。光致發光最常見的類型是熒光(fluorescence)和磷光(phosphorescence)。利用熒光強度進行分析的方法,稱為熒光法(fluorimetry)。在熒光分析中,待測物質分子成為激發態時所吸收的光稱為激發光,處於激發態的分子回到基態時所產生的熒光稱發射光。熒光法測定的是受光激發後所發射的熒光的強弱,而不是測定激發光的強弱。凡能產生熒光的化合的,均可採用熒光分析法進行定性或定量。
(1)熒光強度的影響因素
1)溶劑:增大溶劑的極性,將使π-π※躍遷的能量降低,熒光增強。在水、乙醇、環已烷等這些常用溶劑中常含有熒光雜質,影響測定,必須在使用前作淨化處理。
2)熒光物質的濃度:對於某一熒光物質的稀溶液,在一定頻率和一定強度的放射光Ⅰ。照射下,如果光被吸收的百分率不太大,且溶液的濃度很小,當溶液的厚度不變時,則它所發生的熒光強度F和該溶液的濃度C成正比,即F=φⅠ。ECL式中φ為熒效率。當熒光物質濃度高時,會發生分子間碰撞,使熒光效率降低。因為溫度增高後使分子間碰撞次數增加,消耗分子的內部能量。
3)溫度:大多數情況隨溫度升高時,熒光效率降低。因為溫度增高後使分子間碰撞次數增加,消耗分子的內部能量。
4)溶液的PH值:當熒光物質本身為弱酸或弱鹼時,溶液的pH值改變對溶液熒光強度產生影響較大,因為有些物質在離子狀態時無熒光,而有些則相反,也有二者均有熒光,但熒光光譜有所不同。
(2)熒光定量分析法:熒光定量分析法通常有標準對比法和標準曲線法。操作和計算可參照比色法,但應注意,空白溶液的熒光強度F。往往不在零點,用上述兩種方法時應先測定F,再從標準品Fs和試樣Fx中減去F。後進行計算。
多組分混合物在熒光分析也可根據熒光峰相距情況採取不同的方法,如各組分熒光峰相距頗遠,可分別在不同波長測定各個組分的熒光強度,然後直接求出各組分濃度。如果各組分熒光光譜相互重疊,利用熒光強度的加和性質,在適宜熒光波長處,測得混合物的熒光強度,再根據被測物質各自在適宜波長處的最大熒光強度,列出聯立方程式求算各自的含量。
對較高濃度的熒光物質可用差示熒光法測定。
(3)熒光分析技術的應用:熒光分析法靈敏度高,選擇性好、取樣量少,因此已廣泛應用於各領域,在臨床生化檢驗方面可用於某些無機物與有機物的分析。
無機化合物能直接產生熒光並用於測定的為數不多,但與有機試劑絡合後進行熒光分析的元素已達60餘種,常見無機物的熒光測定見表16-2。
表16-2 常見無機物的熒光測定
| 元素 | 熒光試劑 | 激發波長(nm) | 熒光波長(nm) | 靈敏度(μg/ml) |
| Ag | 四氯熒光素 | 540 | 580 | 0.1 |
| Al | 桑色素 | 430 | 500 | 0.1 |
| Br | 熒光素 | 440 | 470 | 0.002 |
| Ca | 乙二醛-雙-(4-羥苄基腙) | 453 | 523 | 0.0004 |
| Cl | 熒光素+AgNO3 | 254 | 505 | 0.002 |
| CN | 2',7'-雙(乙酸基汞)熒光素 | 500 | 650 | 0.1 |
| Fe | 曙紅+1,10-二氮雜非 | 540 | 580 | 0.1 |
| Pb | 曙紅+1,10-二氮雜非 | 540 | 580 | 0.1 |
| Zn | 8-羥基喹啉 | 365 | 520 | 0.5 |
| F | 石榴茜互R-AI絡合物 | 470 | 500 | 0.001 |
某些有機化合物的熒光測定應用較多,如糖類、胺類、甾族化合物、DNA與RNA、酶與輔酶、維生素等。常見有機化合物的熒光測定法見表16-3。
表16-3 常用有機化合物熒光測定法
| 待測物 | 試劑 | 激發波長(nm) | 熒光波長(nm) | 靈敏度(μg/ml) |
| 核酸 | 溴化乙啶 | 360-365 | 580-590 | 0.1 |
| 蛋白質 | 曙紅y | 紫外 | 540 | 0.06 |
| 胺基酸 | 氧化酶等 | 315 | 425 | 0.01 |
| 腎上腺素 | 乙二胺 | 420 | 525 | 0.001 |
| NAD(P)H | 自身為熒光物質 | 340 | 450 | 10-6mol/L |
| ATP | 已糖激酶、6-磷酸葡萄糖脫氫酶、6-磷酸葡萄糖 | 340 | 450 | 2×10-6mol/L |
| 維生素A | 無水乙醇 | 345 | 490 | 0.001 |
2、火焰光度法火焰光度法是利用火焰中激發態原子回降至基態時發射的光譜強度進行含量分析的方法。它在儀器結構和分析操作上與火焰原子吸收法相似。
在火焰光度法中,試液和助燃氣一起進入霧化室,霧化後噴入火焰,霧粒在火焰中蒸發和激發,激發態原子降落到低能態時發生光輻射,經單色器分光後到達檢測器,然後由顯示系統顯示其發射光強度。
試樣中的待測元素激發態原子的發射光強度I與該元素濃度C成正比關係,即I=aC。式中a為常數。a與試樣的組成、蒸發和激發過程有關。
火焰光度法同樣存在各種因素干擾,如供氣壓力,試樣導入量、有機溶劑和無機酸的影響,以及金屬元素間的相互作用等,某些干擾因素的消除方法同原子吸收法。對於金屬離子間的相互作用可以下述方法予以消除:
(1)陽離子的干擾:第二陽離子的存在可使待測陽離子的電離作用降低而導致以元素形式存在居多,結果發射強度增大,這種現象稱為陽離子增強效應。例如測定鈣時有鉀存在,鉀可抑制鈣的電離,干擾鈣的測定。消除這種干擾的辦法是在標準溶液及試樣中加入本身易電離的金屬如銫和鋰。
(2)陰離子干擾:草酸根、磷酸根和硫酸根可與某些陽離子在火焰溫度下形成僅能緩慢蒸發的化合物而抑制原子激發,結果導致待測元素髮射強度降低。消除這種干擾的辦法是用釋放劑。釋放劑的作用是同干擾陰離子牢固結合,使待測陽離子的激發行為不受干擾,或與待測陽離子形成更穩定而易揮發的配合物。故盡量避免使用磷酸、硫酸、草酸做試劑。
此外,應避免環境污染測試體系。使用的器皿應為塑料製品以防止玻璃器皿中金屬溶出干擾測定。
火焰光度法通常採用的定量方法有標準曲線法、標準加入法和內標法。臨床檢驗工作中前兩種應用較多,而且測定血液或血清中鈉和鉀已成常規。應用火焰光度法測定某些元素的波長及檢測限見表16-4。
表16-4 火焰光度法測定某些元素的波長與檢測限
| 元素 | 測定波長(A) | 檢測限(mg/L) | 元素 | 測定波長(A) | 檢測限(mg/L) |
| Li | 6708 | 10-4 | Ca | 4227 | 3×10-3 |
| Na | 5890 | 10-4 | Sr | 4607 | 0.02 |
| K | 7665 | 10-3 | Ba | 4934 | 0.2 |
| Rb | 7800 | 0.05 | Mg | 3852 | 0.1 |
| Cs | 8521 | 1.0 |
散射光譜分析法主要測定光線通過溶液混懸顆粒後的光吸收或光散射程度的一類定量方法。測定過程與比色法類同,常用法為比濁法。但顆粒的大小和形狀及懸液的穩定性對比濁結果有較大的影響,因此不能完全按比色法的規律進行測定,否則就會引起誤差。
1、儀器和測定方法 由於測定儀器和方法的不同,比濁法又可分為散射測渾法(turbidimetry)和濁度測定法(nephelometry)二類。前者利用一般的光電比色計和分光光度計,其原理是利用光線通過混懸溶液時,由於顆粒的散射使通過的光線減弱,根據光線減弱的程度測定溶液中顆粒的濃度,所以,實際上可將散射測渾法看成是比色分析的一種特殊情況。後者則是直接測定混懸溶液中顆粒散射光的強度,由於一般光電比色計中光源和光電管在一直線上,無法測定散射光線的強度,需要特殊的濁度計,如雷射比濁儀。測定方法可採用速率法或終點法進行。
速率散射比濁法是一種動力學測定方法,1977年由Seternbery首先用於免疫測定,在一定條件下,抗原和相應的抗體很快結合成抗原抗體免疫複合物顆粒,速率比濁法就是在一定時間內抗原抗體結合過程中,測定二者結合的最大反應速度,即反應達頂峰峰值。終點散射比濁法用於免疫測定時,在一定時間內,通常是抗原抗體反應達到平衡,複合物的濁度不再受時間的影響,但必須在聚合形成絮狀沉澱之前進行濁度測定。
2、影響比濁法測定的因素比濁法的突出問題是顆粒的大小對濁度和濁度曲線有較大的影響。因此,在比濁法中必須力求作到:①混懸液中微粒的分散度也就是顆粒的大小應儘可能相同,並易重複。標準管和測定管中顆粒大小應力求一致。②混懸液在一定時間內,至少10分鐘內應維持穩定,也就是顆粒應該不易相互聚集,變粗變大。為此,關鍵在於製備混懸液必須嚴格控制條件,一般應注意;①沉澱劑或抗體的濃度,一般情況下濁度隨其濃度的增高而增大。②混勻方法和速度,一般而言,緩慢加入試劑,逐滴加入,不斷搖勻,易產生粗顆粒沉澱;而迅速加入,迅速搖勻,易產生膠態溶液。③溫度,溫度升高可加快分子間的碰撞,常使某些在室溫細小均勻的顆粒易變為粗大的絮狀沉澱。④pH溶液,pH可影響沉澱的形成及顆粒的大小,如蛋白質、酶類,在其等電點pH值時最易形成顆粒並沉澱。⑤混懸液的穩定性和測定時間,大多數混懸液隨放置時間的延長顆粒變粗而沉澱,吸光度下降,因此應及時比濁。如出現沉澱太快,可以加入保護性膠體,如聚乙烯吡咯烷酮、表面活性劑等,但應注意加入後可能會引起顆粒及光學性質的變化。⑥其它電解質和非電解質存在的干擾。由此可見,比濁法易受外界各種因素的影響,因此在建立一種比濁測定方法時,必須認真探索其反應規律,力求控制各種影響因素以克服比濁法重複性和準確性較差的缺點。
3、比濁法的應用目前在臨床生化檢驗中使用最多的比濁法是免疫比濁法,如免疫球蛋白、載脂蛋白和補體等項目均已大部分用免疫比濁法進行快速定量。
早在1838年Libby等人把比濁法測定就應用到抗原抗體反應上。所謂免疫比濁法是利用抗原和抗體的特異性結合形成複合物,通過測定複合物形成量的多少,對抗原或抗體進行定量的方法。在介質溶液中形成複合物需要一定的條件,包括:①要有特異性抗體。作為組織或體液中蛋白質種類相當多,若要快速特異測定,就需要有單價特異抗體,也就是說,某一種蛋白有其特異抗體才能與該抗原結合,形成免疫複合物,再定量。若抗體不純或混有另一種或兩種少量抗體,這種免疫複合物就不是單一複合物而是大雜燴,結果偏高。②抗原抗體的比例要適當。因免疫複合物的形成有三個階段,第一階段是初步形成抗原抗體二元複合物;第二階段是複合物交聯成大的網路狀結構;第三階段是複合物聚合產生絮狀沉澱。只有在抗原與抗體等價時即無過剩抗體,此時,複合物的結合和解離處在平衡狀態,其混濁程度達高峰,在抗體過量時,隨抗原量的增加而複合物的形成也增加,成正比關係,其測定只能在以應曲線的左側進行。同時,抗原也不能過量,因為抗原過量,則複合物減少,光散射或光吸收就減少,檢測結果偏低。③溶液介質適宜,一般要求溶液中有非離子性親水多聚體促進免疫複合的形成,如聚乙二醇6000等,溶液pH以6.5~8.0之間為宜,離子強度大比小易形成複合物,對離子的種類也有一定的要求。
在免疫比濁過程中,由於抗原與抗體結合有三個階段,從而導致吸光度與濃度之間不呈線性關係,一般是三次程曲線關係。如果要將抗原與抗體兩個變數之間的變動特徵恰當地反映出來,需要經三次程擬合成近似直線化的曲線方程,再進行運算。方法可採用終點法和速率法,用五個不同濃度進行定標,經三次曲線方程求出一條能反映真實情況的濃度與吸光度的關係曲線方程。作為定量的工作曲線。
| 關於「臨床生物化學/光譜分析的常用方法」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |