醫院藥學/藥品生產管理規範
| 醫學電子書 >> 《醫院藥學》 >> 醫院製劑業務管理 >> 藥品生產管理規範 |
| 醫院藥學 |
|
|
GMP(good practice the manfacture and quality control of drugs或稱Good Manufacturing)譯為「優良的生產」或譯為「藥品生產管理規範」。它開始是由美國坦普爾大學6名教授編寫制定的,1963年由美國國會第一次頒布布成為法令。在1969年,第二十二屆世界衛生大會的決議要求所有會員國執行《藥品生產管理規範》。後來,在第二十四屆世界衛生大會上,又作出決定,現成世界衛生組織的理事長繼續研究上述文件中的規定,是否切實可行,並向第二十五屆世界衛生大會作出報告。據此,世界衛生組織現令其專門設置的專家委員會負責,研究各會員國提出的意見,並草擬一個修正方案。這一方案於1974年分發全體會員國,隨後收到約四十個國家的答覆。經專家委員會再次討論最後於1975年第十一月提出了修改後的《藥品生產管理規範》,1977年第二十八屆世界衛生大會討論通過,確定為世界衛生組織的法規。目前世界上已有一百多個國家執行GMP。有一些先進國家還制訂了本國的GMP,GMP分藥品生產的廠房、設備,人員、原材料、工藝規程、生產記錄、生產控制、監控方法、產品包裝、銷售和穩定性等章節。它的基本指導思想是,用全面質量管理,保證藥品的安全性、有效性、穩定性和品質優良,它要求藥品生產的質量管理不限於分析、化驗、檢查表格,車間檢驗和出廠檢驗等,而是涉及整個生產全過程的進行質量監控措施。
(一)GMP的特點
1、它強調藥品生產和質量管理的法律責任。只要生產藥品就要向衛生行政部門登記,就要按GMP要求,接受衛生行政部門的監督。
2、對影響產品質量的因素有嚴格要求,強調生產人員的業務能力、技術水平和教育。
3、強調生產全過程和全面性的質量管理。
4、強調防檢結合,以防為主。
5、廣泛應用數理統計方法。對抽樣和統計的控制限度作了具體規定,以保證樣本的代表性,防止造成無根據的結論。
6、重視為用戶服務。要求建立銷售檔案,保證藥品的穩定性,制訂有效期,建立申訴檔案。
多年的GMP實施結果證明,GMP可以把人為事故降低到最小限度,從根本上保證藥品質量。
(二)GMP的種類
現行的GMP有三種:一是國際的,如WHO提出的GMP;北歐七國自由貿易聰明制定的PIC。二是國家級的,如美國、加拿大、澳大利、日本等都有本國的GMP,它由國家制定,由衛生行政部門監督實施。三是醫藥工業界自定的GMP。
WHO推薦的 GMP是藥品生產和質量管理通用的指南,可使用國家間的投資,合資經營的藥品進出口貿易便利,使相互之間的監督檢查有統一的標準。我國制定GMP後,也便於藥品進出口。
(三)國外GMP概要
1、美國坦普爾大學提出的GMP 坦普爾大學6位教授指出的全面質量管理方案分十五章。第一章定義,第二章成藥,第三章廠房,第四章設備,第五章人員,第六章原材料,第七章工藝流程與生產記錄,第八章生產和監控過程,第九章產品容器,第十章包裝與貼簽,第十一章化驗室管理,第十三章穩定性,第十四章制訂有效期,第十五章申訴檔案。
在第一章中,不僅確定了產品種類和名稱,還提出了物料和文件流動的模式示意圖,見圖12-1。
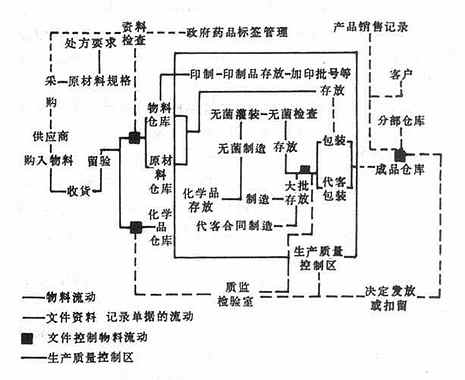
圖12-1 美國GMP物料及文件流動模式圖
2、世界衛生組織公布的GMP 世界衛生組織(WHO)於1979年在第二十八屆世界衛生大會上通過的GMP共分十三章。
第一章概論。其中提到,為保證消費者能獲得高質量的藥品,在生產中實行全面管理是十分重要的。生產藥品時決不允許出現任意的操作方式。優良的制度應被視作通用的準則。
第二章定義。其中提高藥品、生產、批、批號、留驗、質量控制、半成品、起始原料等定義。
第三章人員。其中提到負責指導藥品生產和質量控制的專家,應具備國家法令規定的專門訓練和經驗的資格。他們受的教育應包括:化學;化學工程學;微生物學;藥劑學和技術;藥理學和毒理學;生物學和組織學;其它有關科學。除了上面說的專家以外,還應有一定數量的,經過技術訓練的人員,來進行生產和質量控制。
第四章廠房建築。所有的藥品均應在適合條件的廠房建築里進行生產、加工、包裝、貼簽和試驗。其中提到廠房建築時,要注意與相鄰廠房生產操作的相互影響,要有適當的工作場所,防止混藥、防止污染等問題。
第五章設備。生產用設備必須適合指定用途,便於進行清掃,污染降至最低限度,無菌灌注用的設備,每隔適當階段用微生物學方法加以核查。生產用的量、衡設備,每隔一定階段加以校正和核查。
第六章衛生。所有廠房均應清潔,要求打掃的區域規定每次打掃的期限,需要採取的打掃辦法,指定專人負責清潔衛生工作。在工作區附近,應該有足夠清潔的、通風良好的盥洗設施,包括洗手裝置和更衣室等。
第七章起始原料。藥品生產各個階段所用的一切起始原料都應有所庫存,並應保存關於供應單位、來源、收貨日期、分析日期、質量監督部門批准使用日期和用於生產的日期等。凡是被接受或批准的起始原料均應有妥善和明顯的標誌。所有無法使用的起始原料均應有明顯標誌,並應忙地予以處理或退回供應單位。
第八章生產操作。所有生產操作和控制都應在專家指導下進行。操作要特別注意:清潔工作、設備容器、污染和混雜的預防、滅菌操作的特殊要求、抗生素類生產的特殊要求等;對生產人員要求:不能患有傳染病骸身體裸露、表面患有開放性疾病,工作服、鞋帽不能臟污等;對與生產程序有關文件的制訂、生產批次記錄等。其物料文件流程要求如圖12-1。
第九章標籤和包裝。標籤和包裝材料,包括說明書在內,必須按不同的產品分別儲存和處理,只有經過授權的人員方能接觸這種材料。標籤和包裝材料應包括書籍發放數量、包裝和貼簽後應該計數量。所有已經編號而未用的標籤應全部銷毀。
第十章質量監控制度。每一個生產單位都應有一個質量監督部門,在有適宜資格的專家指導下進行工作,直接對總管理負責,而獨立於其它的部門。質量監督部門必須控制所有的起始原料,監查生產操作中有關質量部分,並對藥品質量和穩定性實行監督。質量監督部門必須有一個專用的實驗室。
第十一章自檢。為了能嚴格的與所有生產程序及規定的控制要求相符合,製藥企業可考慮指定一個專家或一批專家,負責經常地對它的整個生產及控制操作進行定期檢查。
第十二章發送記錄。對於已經製成的每一批藥品,都應保存關於發送情況的合適記錄,以便於必要時能迅速而全部地將這批藥品撤回。
第十三章關於不良反應的報告和控訴。關於用藥後引起損害或不良反應的報告,必須送交適當的負責人審閱,有關藥品質量的控訴,必須徹底地進行查究。如果證明這些控訴確有根據,應儘可能迅速地採取適當措施。已經採取的措施必須記錄下來,並與原來的控訴一起歸檔。
| 關於「醫院藥學/藥品生產管理規範」的留言: | |
|
目前暫無留言 | |
| 添加留言 | |